metal-organic papers
Acta Cryst.(2006). E62, m87–m89 doi:10.1107/S1600536805040341 Denget al. [Cd(C
6H2O4S)(C3H4N2)2(H2O)]
m87
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
catena
-Poly[[aquadiimidazolecadmium(II)]-l
-thiophene-2,5-dicarboxylato]
Zhao-Peng Deng, Shan Gao,* Li-Hua Huo and Hui Zhao
Laboratory of Functional Materials, School of Chemistry and Materials Science, Heilongjiang University, Harbin 150080, People’s Republic of China
Correspondence e-mail: shangao67@yahoo.com
Key indicators
Single-crystal X-ray study
T= 295 K
Mean(C–C) = 0.014 A˚
Rfactor = 0.041
wRfactor = 0.168
Data-to-parameter ratio = 16.7
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2006 International Union of Crystallography Printed in Great Britain – all rights reserved
The CdIIatom has distorted pentagonal bipyramidal coord-ination geometry in the coordcoord-ination polymer [Cd(TDA)-(Him)2(H2O)]n [where TDA
2
is the thiophene-2,5-dicarboxylate dianion (C6H2O4S
2
) and Him is imidazole (C3H4N2)]. The cadmium ion is bound by four carboxylate O
atoms from two independent TDA2 groups, two N atoms from two different imidazole ligands, and one water molecule. The carboxylate groups bind in bidentate mode to the Cd center, forming a linear chain structure such that the closest Cd Cd distance is 10.577 (6) A˚ . The polymeric chains are connectedviahydrogen bonds and–stacking interactions into a three-dimensional network.
Comment
Thiophene-2,5-dicarboxylic acid (H2TDA) is reported to be a
potential anticancer agent (Sahasrabudhe et al., 1960), and additionally an excellent building block for constructing coordination polymers (Chen et al., 1993). In previously studied polymers of this type, the versatile H2TDA ligand
shows a variety of binding modes to metal ions, from mono- to tetradentate (Chenet al., 1998, 1999). In order to further the study of H2TDA coordination modes, we report here the
synthesis and crystal structure of the title CdII coordination polymer, [Cd(TDA)(Him)2(H2O)]n, (I). It was obtained by the hydrothermal reaction of cadmium dinitrate tetrahydrate, thiophene-2,5-dicarboxylic acid and imidazole (Him) in an aqueous solution.
As illustrated in Fig. 1, the asymmetric unit of (I) consists of one CdIIion, one TDA2dianion, two imidazole ligands and
one coordinated water molecule. Each CdII atom is
seven-coordinate and bound by four carboxylate O atoms from two independent TDA2groups, two N atoms from two different imidazole ligands, and one water molecule. The local coord-ination of the CdII atom can be described as distorted pentagonal bipyramidal. The equatorial pentagonal plane is
defined by atoms O1, O2, O3i, O4iand N1 [symmetry code: (i)
x,y1,z]. Atom N3 and the water molecule occupy the axial sites. The smallest O—Cd—O angle, 49.1 (3), is attributed to
the bis-chelate coordination of the TDA2ligand that forms two four-membered rings. It should be noted that the two C— O bond distances of the carboxylate group (O3/C6/O4) are almost equivalent, and so in agreement with its delocalized state, whereas the O2—C1 distance is longer than the O1—C1 distance, in accordance with the formal double-bond character of the O1—C1 bond. The dihedral angles between the two carboxylate groups and the thiophene ring are 5.3 (4) (O1/
C1/O2) and 6.0 (4) (O3/C6/O4), respectively, demonstrating
that the TDA2ligand is basically planar.
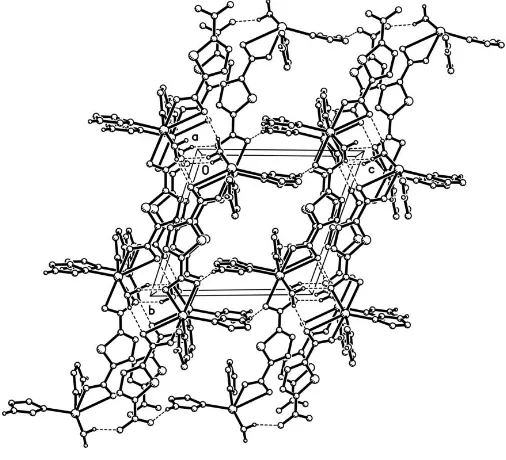
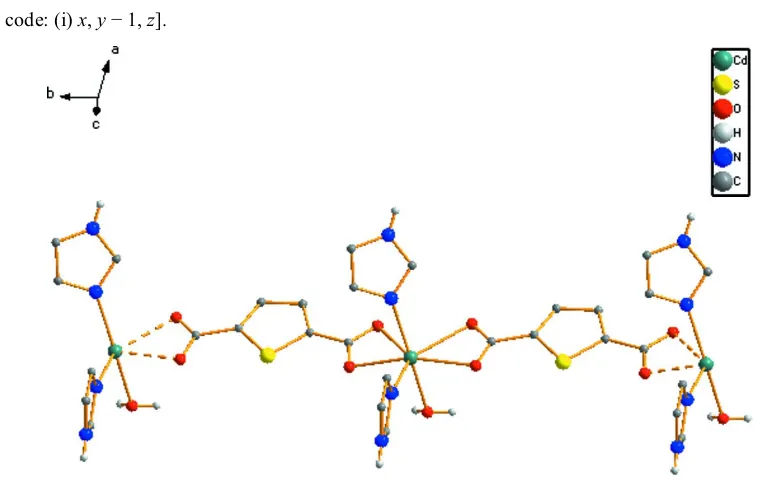
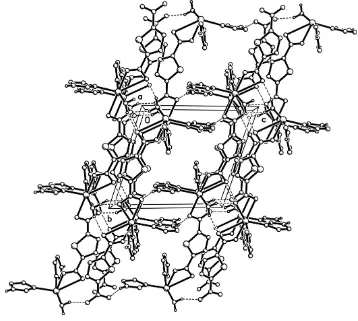
Each TDA2group binds in bis-bidentate chelating mode to link neighboring CdII atoms to form a one-dimensional linear chain structure which is propagated parallel to thebaxis (Fig. 2), in which the closest Cd Cd distance is 10.577 (6) A˚ . Furthermore, the water and imidazole molecules form extensive intermolecular hydrogen bonds with carboxylate O atoms (Table 2). There are–stacking interactions between adjacent thiophene rings, the centroid–centroid separation being 3.694 (6) A˚ . The polymeric chains align in a manner that
facilitates both hydrogen-bonding and – stacking inter-actions, leading to a three-dimensional supramolecular network (Fig. 3).
Experimental
Cadmium dinitrate tetrahydrate (3.08 g, 10 mmol) was added to an aqueous solution of thiophene-2,5-dicarboxylic acid (1.72 g, 10 mmol). The pH was adjusted to 6 with 0.1Msodium hydroxide. Imidazole (1.34 g, 20 mmol) was then added. The mixture was stirred for 1 h and then sealed in a 50 ml Teflon-lined stainless steel bomb and held at 383 K for 3 d. The bomb was cooled naturally to room temperature, and colorless prismatic crystals of (I) were obtained. Analysis calculated for C12H12N4O5SCd: C 33.00, H 2.77, N 12.83%;
found: C 32.96, H 2.74, N 12.85%.
Crystal data
[Cd(C6H2O4S)(C3H4N2)2(H2O)]
Mr= 436.72 Triclinic,P1
a= 7.7486 (15) A˚
b= 10.577 (2) A˚
c= 10.947 (2) A˚
= 103.71 (3)
= 101.57 (3)
= 108.43 (3)
V= 789.1 (4) A˚3
Z= 2
Dx= 1.838 Mg m3
MoKradiation Cell parameters from 7316
reflections
= 3.0–27.5
= 1.55 mm1
T= 295 (2) K Prism, colorless 0.360.240.18 mm
Data collection
Rigaku R-AXIS RAPID diffractometer
!scans
Absorption correction: multi-scan (ABSCOR; Higashi, 1995)
Tmin= 0.641,Tmax= 0.761
7628 measured reflections
3564 independent reflections 2125 reflections withI> 2(I)
Rint= 0.086 max= 27.5
h=9!10
k=13!13
l=14!11
metal-organic papers
m88
Denget al. [Cd(C6H2O4S)(C3H4N2)2(H2O)] Acta Cryst.(2006). E62, m87–m89 Figure 2
[image:2.610.46.299.72.230.2]The linear chain structure of the title complex. H atoms attached to C atoms have been omitted.
Figure 3
[image:2.610.313.566.73.303.2]Packing diagram of the title complex, viewed along the a axis. The hydrogen bonds are shown as dashed lines. H atoms attached to C atoms have been omitted.
Figure 1
ORTEPIIplot (Johnson, 1976) of the title complex, with displacement ellipsoids drawn at the 30% probability level. [Symmetry code: (i)x,y
[image:2.610.45.299.283.441.2]Refinement
Refinement onF2 R[F2> 2(F2)] = 0.041
wR(F2) = 0.168
S= 1.06 3564 reflections 214 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2(F
o2) + (0.0795P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 1.78 e A˚
3
min=1.65 e A˚
[image:3.610.43.298.191.354.2]3
Table 1
Selected geometric parameters (A˚ ,).
Cd1—O1w 2.341 (6) Cd1—O1 2.829 (6) Cd1—O2 2.318 (6) Cd1—O3i
2.503 (6) Cd1—O4i
2.413 (6) Cd1—N1 2.267 (8)
Cd1—N3 2.269 (7) O1—C1 1.227 (14) O2—C1 1.249 (14) O3—C6 1.244 (9) O4—C6 1.248 (10) N3—Cd1—O1 80.4 (3)
O1w—Cd1—O1 86.1 (3) O2—Cd1—O1 49.1 (3) O1—Cd1—O3i
84.4 (3) O1—Cd1—O4i
136.0 (2) O1—Cd1—N1 139.2 (2) N1—Cd1—N3 96.3 (3) N1—Cd1—O2 90.5 (3) N3—Cd1—O2 90.2 (2) N1—Cd1—O1w 95.8 (2) N3—Cd1—O1w 166.2 (2)
O2—Cd1—O1w 83.2 (2) N1—Cd1—O4i 84.6 (3) N3—Cd1—O4i 103.5 (2) O2—Cd1—O4i 165.8 (2) O1w—Cd1—O4i
84.1 (2) N1—Cd1—O3i 136.3 (2) N3—Cd1—O3i
86.4 (2) O2—Cd1—O3i
133.2 (2) O1w—Cd1—O3i
89.3 (2) O4i
—Cd1—O3i
52.7 (2)
Symmetry code: (i)x;y1;z.
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O1w—H1w1 O3ii
0.85 (7) 1.93 (4) 2.732 (8) 157 (8) O1w—H1w2 O1iii
0.85 (6) 1.96 (3) 2.770 (10) 160 (7) N2—H14 O4iv 0.86 1.92 2.758 (10) 165 N4—H15 O2v
0.86 1.97 2.826 (10) 171
Symmetry codes: (ii) x;yþ1;z; (iii)x;y;z; (iv)x;yþ1;zþ1; (v)
xþ1;y;z.
The H atoms attached to C atoms and imidazole N atoms were placed in calculated positions, with C—H = 0.93 A˚ , N—H = 0.86 A˚ andUiso(H) = 1.2Ueq(C,N), and were refined with the riding-model
approximation. Water H atoms were located in a difference Fourier map and refined with O—H and H H distance restraints of 0.85 (1) and 1.39 (1) A˚ , respectively, and with Uiso(H) = 1.5Ueq(O). The
largest residual peak is 1.08 A˚ from the Cd atom and the deepest is hole is 0.9 A˚ from the same atom.
Data collection:RAPID-AUTO(Rigaku Corporation, 1998); cell refinement: RAPID-AUTO; data reduction: CrystalStructure
(Rigaku/MSC, 2002); program(s) used to solve structure:SHELXS97
(Sheldrick, 1997); program(s) used to refine structure:SHELXL97
(Sheldrick, 1997); molecular graphics: ORTEPII (Johnson, 1976); software used to prepare material for publication:SHELXL97.
The authors thank the National Natural Science Foundation of China (No. 20101003), the Scientific Fund of Remarkable Teachers of Heilongjiang Province (1054 G036) and Heilongjiang University for supporting this study.
References
Chen, B. L., Mok, K. F., Ng, S. C. & Drew, M. G. B. (1999).Polyhedron,18, 1211–1220.
Chen, B. L., Mok, K. F., Ng, S. C., Feng, Y. L. & Liu, S. X. (1998).Polyhedron,
17, 4237–4247.
Chen, C. T. & Suslick, K. S. (1993).Coord. Chem. Rev.128, 293–322. Higashi, T. (1995).ABSCOR. Rigaku Corporation, Tokyo, Japan.
Johnson, C. K. (1976).ORTEPII. Report ORNL-5138, Oak Ridge National Laboratory, Tennessee, USA.
Rigaku Corporation (1998). RAPID-AUTO. Rigaku Corporation, Tokyo, Japan.
Rigaku/MSC (2002). CrystalStructure. Rigaku/MSC Inc., 9009 New Trails Drive, The Woodlands, TX 77381-5209, USA.
Sahasrabudhe, M. B., Nerurkar, M. K., Nerurkar, M. V., Tilak, B. D. & Bhavasar, M. D. (1960).Br. J. Cancer.14, 547–554.
Sheldrick, G. M. (1997). SHELXL97 and SHELXS97. University of Go¨ttingen, Germany.
metal-organic papers
Acta Cryst.(2006). E62, m87–m89 Denget al. [Cd(C
supporting information
sup-1
Acta Cryst. (2006). E62, m87–m89supporting information
Acta Cryst. (2006). E62, m87–m89 [doi:10.1107/S1600536805040341]
catena
-Poly[[aquadiimidazolecadmium(II)]-
µ
-thiophene-2,5-dicarboxylato]
Zhao-Peng Deng, Shan Gao, Li-Hua Huo and Hui Zhao
S1. Comment
Thiophene-2,5-dicarboxylic acid (H2TDA) is reported to be a potential anticancer agent (Sahasrabudhe et al., 1960), and
additionally an excellent building block for constructing coordination polymers (Chen et al., 1993). In previously studied polymers of this type, the versatile H2TDA ligand shows a variety of binding modes to metal ions, from mono- to
tetradentate (Chen et al., 1998, 1999). In order to further the study of H2TDA coordination modes, we report here the
synthesis and crystal structure of the title CdII coordination polymer, [Cd(TDA)(Him)
2(H2O)]n, (I). It was obtained by the
hydrothermal reaction of cadmium dinitrate tetrahydrate, thiophene-2,5-dicarboxylic acid and imidazole (Him) in an aqueous solution.
As illustrated in Fig. 1, the asymmetric unit of (I) consists of one CdII ion, one TDA2− dianion, two imidazole ligands
and one coordinated water molecule. Each CdII atom is seven-coordinate and bound by four carboxyl O atoms from two
independent TDA2− groups, two N atoms from two different imidazole ligands, and one water molecule. The local
coordination sphere of the CdII atom can be described as distorted pentagonal bipyramidal. The equatorial pentagonal
plane is defined by atoms O1, O2, O3i, O4i and N1 [symmetry code: (i) x, y − 1, z]. Atom N3 and the water molecule
occupy the axial sites. The smallest O—Cd—O angle, 49.1 (3)°, is attributed to the bis-chelate coordination of the TDA2−
ligand that forms two four-membered rings. It should be noted that the two C—O bond distances of the carboxyl group (O3/C6/O4) are almost equivalent, and so in agreement with its delocalized state, whereas the O2—C1 distance is longer than the O1—C1 distance, in accordance with the formal double-bond character of the O1—C1 bond. The dihedral angles between the two carboxyl groups and the thiophene ring are 5.3 (4)° (O1/C1/O2) and 6.0 (4)° (O3/C6/O4), respectively, demonstrating that the TDA2− ligand is basically planar.
Each TDA2− group binds in bis-bidentate chelating mode to link neighbouring CdII atoms to form a one-dimensional
linear chain structure which is propagated parallel to the b axis (Fig. 2), in which the closest Cd···Cd distance is 10.577 (6) Å. Furthermore, the water and imidazole molecules form extensive intermolecular hydrogen bonds with carboxylate O atoms (Table 2). There are π–π stacking interactions between adjacent thiophene rings, the centroid– centroid separation being 3.694 (6) Å. The polymeric chains align in a manner that facilitates both hydrogen-bonding and
π–π stacking interactions, leading to a three-dimensional supramolecular network (Fig. 3).
S2. Experimental
supporting information
sup-2
Acta Cryst. (2006). E62, m87–m89S3. Refinement
The H atoms attached to C atoms and imidazole N atoms were placed in calculated positions, with C—H = 0.93 Å, N—H = 0.86 Å and Uiso(H) = 1.2Ueq(C,N), and were refined with the riding-model approximation. Water H atoms were located
in a difference Fourier map and refined with O—H and H···H distance restraints of 0.85 (1) and 1.39 (1) Å, respectively, and with Uiso(H) = 1.5Ueq(O). The largest residual peak is 1.08 Å from the Cd atom and the deepest is hole is ?? Å from
[image:5.610.128.484.172.391.2]??.
Figure 1
ORTEPII plot (Johnson, 1976) of the title complex, with displacement ellipsoids drawn at the 30% probability level.
[Symmetry code: (i) x, y − 1, z].
Figure 2
[image:5.610.105.492.424.670.2]supporting information
[image:6.610.124.482.72.393.2]sup-3
Acta Cryst. (2006). E62, m87–m89Figure 3
Packing diagram of the title complex along the a axis. The hydrogen bonds are shown as dashed lines. H atoms attached to C atoms have been omitted.
catena-Poly[[aquadiimidazolecadmium(II)]-µ-thiophene-2,5-dicarboxylato]
Crystal data
[Cd(C6H2O4S)(C3H4N2)2(H2O)] Mr = 436.72
Triclinic, P1 Hall symbol: -P 1
a = 7.7486 (15) Å
b = 10.577 (2) Å
c = 10.947 (2) Å
α = 103.71 (3)°
β = 101.57 (3)°
γ = 108.43 (3)°
V = 789.1 (4) Å3
Z = 2
F(000) = 432
Dx = 1.838 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 7316 reflections
θ = 3.0–27.5°
µ = 1.55 mm−1 T = 295 K Prism, colorless 0.36 × 0.24 × 0.18 mm
Data collection
Rigaku R-AXIS RAPID diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 10 pixels mm-1 ω scans
Absorption correction: multi-scan
(ABSCOR; Higashi, 1995)
Tmin = 0.641, Tmax = 0.761
7628 measured reflections 3564 independent reflections 2125 reflections with I > 2σ(I)
supporting information
sup-4
Acta Cryst. (2006). E62, m87–m89θmax = 27.5°, θmin = 3.0° h = −9→10
k = −13→13
l = −14→11
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.041 wR(F2) = 0.168 S = 1.06 3564 reflections 214 parameters 3 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0795P)2]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 1.78 e Å−3
Δρmin = −1.65 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
supporting information
sup-5
Acta Cryst. (2006). E62, m87–m89H12 0.8920 0.5387 0.4004 0.072* H14 −0.0026 0.1686 0.6627 0.061* H15 0.9317 0.3296 0.2835 0.065* H1W1 −0.142 (14) 0.049 (8) 0.075 (6) 0.062* H1W2 −0.132 (14) −0.073 (3) 0.096 (7) 0.062* H3 0.3255 0.4714 −0.0639 0.053* H4 0.3555 0.7179 −0.0374 0.042* H7 −0.0854 0.1441 0.4419 0.057*
H8 0.4618 0.2406 0.5683 0.082*
H9 0.3344 0.2389 0.7495 0.076*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Cd1 0.0388 (3) 0.0268 (3) 0.0415 (4) 0.0160 (2) 0.0178 (2) 0.0159 (2) S1 0.0380 (11) 0.0313 (11) 0.0513 (13) 0.0172 (9) 0.0196 (9) 0.0236 (10) O1W 0.047 (3) 0.034 (3) 0.041 (3) 0.014 (3) 0.011 (3) 0.012 (3) O1 0.068 (5) 0.021 (4) 0.146 (8) 0.025 (3) 0.014 (5) 0.007 (4) O2 0.050 (4) 0.040 (4) 0.116 (6) 0.027 (3) 0.036 (4) 0.050 (4) O3 0.059 (4) 0.028 (3) 0.058 (4) 0.021 (3) 0.018 (3) 0.023 (3) O4 0.066 (4) 0.042 (4) 0.061 (4) 0.028 (3) 0.035 (3) 0.023 (3) N1 0.043 (4) 0.061 (5) 0.046 (4) 0.012 (4) 0.020 (3) 0.025 (4) N2 0.082 (6) 0.042 (5) 0.046 (5) 0.033 (4) 0.041 (4) 0.015 (4) N3 0.037 (4) 0.034 (4) 0.062 (5) 0.015 (3) 0.019 (3) 0.017 (4) N4 0.032 (4) 0.061 (6) 0.080 (6) 0.021 (4) 0.018 (4) 0.036 (5) C1 0.026 (4) 0.026 (5) 0.122 (10) 0.013 (4) 0.000 (5) 0.027 (6) C2 0.026 (4) 0.033 (5) 0.056 (6) 0.009 (3) 0.004 (4) 0.015 (4) C3 0.050 (5) 0.036 (5) 0.041 (5) 0.019 (4) 0.011 (4) 0.000 (4) C4 0.053 (5) 0.025 (4) 0.034 (4) 0.024 (4) 0.016 (4) 0.006 (3) C5 0.031 (4) 0.023 (4) 0.032 (4) 0.010 (3) 0.006 (3) 0.010 (3) C6 0.032 (4) 0.026 (4) 0.037 (4) 0.009 (3) 0.006 (3) 0.009 (4) C7 0.049 (5) 0.036 (5) 0.067 (7) 0.023 (4) 0.020 (5) 0.021 (5) C8 0.040 (5) 0.094 (9) 0.039 (6) −0.002 (5) 0.004 (4) 0.008 (6) C9 0.074 (7) 0.067 (8) 0.032 (5) 0.020 (6) 0.017 (5) −0.004 (5) C10 0.042 (5) 0.040 (5) 0.064 (6) 0.025 (4) 0.018 (4) 0.023 (5) C11 0.072 (7) 0.043 (6) 0.060 (7) 0.010 (5) 0.035 (5) −0.011 (5) C12 0.051 (6) 0.041 (6) 0.072 (7) 0.007 (5) 0.014 (5) 0.007 (5)
Geometric parameters (Å, º)
Cd1—O1W 2.341 (6) N2—H14 0.8600
Cd1—O1 2.829 (6) N3—C10 1.326 (11)
Cd1—O2 2.318 (6) N3—C11 1.355 (12)
Cd1—O3i 2.503 (6) N4—C10 1.346 (12)
Cd1—O4i 2.413 (6) N4—C12 1.356 (13)
Cd1—N1 2.267 (8) N4—H15 0.8600
Cd1—N3 2.269 (7) C1—C2 1.489 (12)
supporting information
sup-6
Acta Cryst. (2006). E62, m87–m89O2—C1 1.249 (14) C3—C4 1.444 (12)
O3—C6 1.244 (9) C3—H3 0.9300
O4—C6 1.248 (10) C4—C5 1.374 (11)
S1—C2 1.716 (10) C4—H4 0.9300
S1—C5 1.716 (7) C5—C6 1.485 (11)
O1W—H1W1 0.85 (7) C7—H7 0.9300
O1W—H1W2 0.85 (6) C8—C9 1.306 (14)
O3—Cd1ii 2.503 (6) C8—H8 0.9300
O4—Cd1ii 2.413 (6) C9—H9 0.9300
N1—C7 1.340 (11) C10—H10 0.9300
N1—C8 1.387 (12) C11—C12 1.329 (14)
N2—C7 1.277 (12) C11—H11 0.9300
N2—C9 1.333 (13) C12—H12 0.9300
N3—Cd1—O1 80.4 (3) N4—C12—H12 127.5 O1W—Cd1—O1 86.1 (3) C1—C2—S1 120.7 (8) O2—Cd1—O1 49.1 (3) C1—O2—Cd1 104.5 (6) O1—Cd1—O3i 84.4 (3) C2—C3—C4 109.7 (8)
O1—Cd1—O4i 136.0 (2) C2—C3—H3 125.2
O1—CD1—N1 139.2 (2) C2—S1—C5 92.8 (4) N1—Cd1—N3 96.3 (3) C3—C2—C1 126.7 (9) N1—Cd1—O2 90.5 (3) C3—C2—S1 112.6 (6)
N3—Cd1—O2 90.2 (2) C3—C4—H4 122.7
N1—Cd1—O1W 95.8 (2) C4—C3—H3 125.2 N3—Cd1—O1W 166.2 (2) C4—C5—C6 128.1 (7) O2—Cd1—O1W 83.2 (2) C4—C5—S1 110.4 (6) N1—Cd1—O4i 84.6 (3) C5—C4—C3 114.6 (8)
N3—Cd1—O4i 103.5 (2) C5—C4—H4 122.7
O2—Cd1—O4i 165.8 (2) C6—C5—S1 121.4 (6)
O1W—Cd1—O4i 84.1 (2) C6—O3—Cd1ii 90.4 (5)
N1—Cd1—O3i 136.3 (2) C6—O4—Cd1ii 94.5 (5)
N3—Cd1—O3i 86.4 (2) C7—N1—C8 101.7 (8)
O2—Cd1—O3i 133.2 (2) C7—N1—Cd1 132.8 (7)
O1W—Cd1—O3i 89.3 (2) C7—N2—C9 107.0 (8)
O4i—Cd1—O3i 52.7 (2) C7—N2—H14 126.5
Cd1—O1W—H1W1 108 (7) C8—C9—H9 125.9 Cd1—O1W—H1W2 108 (7) C8—C9—N2 108.2 (9) O1—C1—C2 117.1 (12) C8—N1—Cd1 125.1 (7)
O1—C1—O2 124.2 (9) C9—C8—H8 125.2
O2—C1—C2 118.7 (10) C9—C8—N1 109.5 (9) O3—C6—C5 119.4 (8) C9—N2—H14 126.5 O3—C6—O4 122.4 (8) C10—N3—C11 105.6 (8) O4—C6—C5 118.3 (7) C10—N3—Cd1 126.6 (7) N1—C7—H7 123.3 C10—N4—C12 108.8 (8)
N1—C8—H8 125.2 C10—N4—H15 125.6
N2—C7—H7 123.3 C11—C12—H12 127.5
supporting information
sup-7
Acta Cryst. (2006). E62, m87–m89N3—C10—H10 125.5 C12—C11—H11 124.4 N3—C10—N4 109.0 (9) C12—C11—N3 111.2 (9)
N3—C11—H11 124.4 C12—N4—H15 125.6
N4—C10—H10 125.5 H1W1—O1W—H1W2 109 (7)
C1—C2—C3—C4 −177.8 (7) N1—Cd1—O2—C1 166.5 (6) C10—N3—C11—C12 −4.8 (13) N3—C11—C12—N4 4.4 (14) C10—N4—C12—C11 −2.3 (12) N3—Cd1—N1—C7 164.2 (9) C11—N3—C10—N4 3.2 (11) N3—Cd1—N1—C8 −23.8 (9) C12—N4—C10—N3 −0.6 (11) N3—Cd1—O2—C1 70.2 (6) C2—C3—C4—C5 −1.1 (10) O1—C1—C2—C3 −3.9 (13) C2—S1—C5—C4 −0.8 (6) O1—C1—C2—S1 177.9 (7) C2—S1—C5—C6 177.1 (6) O1W—Cd1—N1—C7 −9.2 (9) C3—C4—C5—C6 −176.5 (7) O1W—Cd1—N1—C8 162.7 (9) C3—C4—C5—S1 1.2 (9) O1W—Cd1—N3—C10 −76.2 (13) C4—C5—C6—O3 4.1 (12) O1W—Cd1—N3—C11 86.0 (14) C4—C5—C6—O4 −176.0 (8) O1W—Cd1—O2—C1 −97.7 (6) C5—S1—C2—C1 178.6 (6) O2—C1—C2—C3 174.1 (8) C5—S1—C2—C3 0.1 (6) O2—C1—C2—S1 −4.2 (11) C7—N1—C8—C9 2.9 (13) O2—Cd1—N1—C7 74.0 (9) C7—N2—C9—C8 −2.4 (13) O2—Cd1—N1—C8 −114.1 (9) C8—N1—C7—N2 −4.6 (12) O2—Cd1—N3—C10 −137.3 (8) C9—N2—C7—N1 4.6 (11) O2—Cd1—N3—C11 24.9 (9) Cd1—N1—C7—N2 168.7 (7) O3i—Cd1—N1—C7 −104.3 (9)
Cd1—N1—C8—C9 −171.1 (8) O3i—Cd1—N1—C8 67.6 (10)
Cd1—N3—C10—N4 168.3 (6) O3i—Cd1—N3—C10 −4.0 (8)
Cd1—N3—C11—C12 −170.1 (8) O3i—Cd1—N3—C11 158.2 (9)
Cd1—O2—C1—C2 −163.0 (6) O3i—Cd1—O2—C1 −15.1 (7)
Cd1—O2—C1—O1 14.8 (11) O4i—Cd1—N1—C7 −92.7 (9)
Cd1ii—O3—C6—C5 179.6 (6) O4i—Cd1—N1—C8 79.2 (9)
Cd1ii—O3—C6—O4 −0.3 (8) O4i—Cd1—N3—C10 46.2 (8)
Cd1ii—O4—C6—C5 −179.6 (6) O4i—Cd1—N3—C11 −151.6 (8)
Cd1ii—O4—C6—O3 0.3 (8) O4i—Cd1—O2—C1 −123.9 (10)
N1—C8—C9—N2 −0.4 (14) S1—C2—C3—C4 0.5 (9) N1—Cd1—N3—C10 132.2 (8) S1—C5—C6—O3 −173.4 (6) N1—Cd1—N3—C11 −65.6 (9) S1—C5—C6—O4 6.5 (10)
Symmetry codes: (i) x, y−1, z; (ii) x, y+1, z.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O1W—H1W1···O3iii 0.85 (7) 1.93 (4) 2.732 (8) 157 (8)
O1W—H1W2···O1iv 0.85 (6) 1.96 (3) 2.770 (10) 160 (7)
N2—H14···O4v 0.86 1.92 2.758 (10) 165
N4—H15···O2vi 0.86 1.97 2.826 (10) 171