Volume 8, No. 5, May – June 2017
International Journal of Advanced Research in Computer Science
RESEARCH PAPER
Available Online at www.ijarcs.info
Comparison of Different Sequence Alignment Methods- A Survey
Yadvir KaurStudent, Department of Computer Engineering, Punjabi University, Patiala,
Punjab, India.
Neelofar Sohi
Assistant Professor, Department of Computer Engineering, Punjabi University, Patiala,
Punjab, India.
Abstract: Bioinformatics is a promising and inventive research field. Biological Sequence alignment is the inborn part of bioinformatics, which helps to find similarity between biological sequences i.e. DNA and protein. Alignment of biological sequences helps to discover functional and structural similarity of sequences. The biological sequence database has been expanding rapidly due to new sequences being found, which has raised the demand to employ more efficient and fast algorithm. There has been an eruption algorithm in the past few decades to find optimal or nearly-optimal alignments. This paper is focused on the popular sequence alignment algorithms. Different types of alignment method have been discussed on the basis of their optimality and approximate solutions. It has been studied that optimal algorithms, which are based on dynamic programming are giving exact solutions. But these are highly computationally complexed. The stochastic optimization methods has been chosen from literature as the potential candidates for the solution of complex multiple sequence alignments with better speed and care.
Keywords: bioinformatics; sequence alignment; DNA; RNA; optimal alignment methods.
I. INTRODUCTION
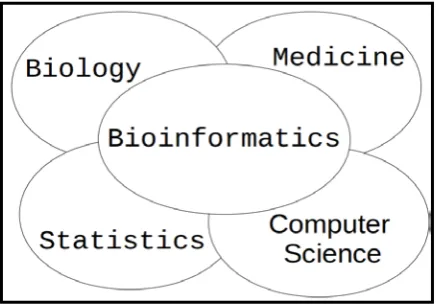
Bioinformatics is an interdisciplinary research area at the interface between biology, computer science, medicine and statistics as shown in Fig. 1. It is a union of biology and informatics, as it involves the computers techniques for storage, retrieval and manipulation of information related to biomolecules for example, Deoxyribonucleic acid (DNA), Ribonucleic acid (RNA) and proteins.
Figure 1. Bioinformatics – An interdisciplinary field.
Sequence alignment is an active research area under bioinformatics. Sequence alignment compares two or more sequences to align their residues (e.g., nucleotide bases of DNA and RNA, or amino acids of a protein). The optimal alignment procedure arranges sequences in such a way so as to maximize the number of identical residue matches. The unaligned and aligned sequences are shown in Fig. 2.
There are various purposes behind the sequence alignment task. Sequence alignment is an important step as it helps to discover structural, operational and evolutionary relationship between the aligned sequences. Biologists work with these aligned sequences to build phylogenetic trees, characterize protein families, and foresee protein structure. The analysis of sequences has helped biologists to detect pathogens, develop drugs, and identify common genes.
The vast amount of biological data that is stored in the form of DNA, RNA and protein sequences requires extensive processing power to retrieve and analyse sequences quickly and precisely. With new biological sequences being found almost on an everyday premise, the biological sequence database is developing exponentially. This explosion of data demands new algorithms which are quick but then proficient. There has been a blast of new algorithms, of which famous algorithms are examined in this paper.
A. Biological Sequences
Biological sequence is either a DNA, ribonucleic acid (RNA), or amino acid (protein) sequence.
DNA/RNA are constituted of nucleotide bases. The nucleotide bases are: adenine (A), thymine (T), cytosine (C), guanine (G), and uracil (U). On the other hand, protein are constituted of amino acids. DNA, RNA or protein sequence or string consists of their respective alphabets as shown below:
DNA (4 bases) : {A,C, G,T} RNA (4 bases) : {A,C,G,U}
[image:1.595.51.271.438.591.2]Proteins (20 amino acids): {A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y}.
B. Sequence Alignment
Sequence alignment finds maximum regions of similarity between two or more DNA or amino acid sequences. It may help to provide additional information such as functional, structural, evolutionary relationships between the sequences. On the basis of similarity in two DNA, RNA or amino acid sequences, there is a chance, that they are homologous i.e. share a common ancestral sequence and their relative differences are the result of mutations.
The mutation is a change in our DNA sequence, that may result from substitution where one symbol is replaced by another, insertion where a new symbol is inserted into the sequence or detention where a symbol is removed.
The aligned residues arranged as parallel rows can be a match, a mismatch, or a gap. To handle insertions and deletions (indel) events, the notion of gaps (Marked by the symbol ‘- ’) is used and are assigned penalties in the scoring process. The alignment score is the sum of the values given to each column [1].
(a) (b)
Figure 3. (a)Global alignment, (b) Local alignment.
Methods of sequence alignment are pairwise sequence alignment and multiple sequence alignment. Pairwise sequence alignment is an alignment between any two given sequences, whereas multiple sequence alignment is an extension of pairwise alignment and involves more than two sequences at a time.
The sequence alignment problem has 2 computational approaches: local alignment and global alignment [2], which are shown in Fig 3. In global alignment, Global sequence alignment attempts to match as many characters as possible, from beginning to the end of both the sequences to find the best possible alignment, whereas Local sequence alignments attempts to identify local regions with the highest level of similarity within the two given sequences.
Figure 4. PAM250 Substitution Matrix [6].
Scoring measures should be applied to decide how the sequences are similar. A scoring framework incorporates scores for the events: matches, mismatches, insertions, deletions and substitutions, In useful terms it can be split into two segments: substitution matrices and gap penalties.
A substitution matrix has dimensions 20 x 20 for proteins and 4 x 4 for DNA. For DNA sequences, a straightforward matrix usually utilized assigns a positive value for a match and a negative value for a mismatch. For protein sequences the well-known matrices are PAM [3] (percent of accepted mutation), which is shown in Fig. 4 and BLOSUM [4] (blocks of amino acid substitution matrices), which is shown in Fig. 5.
[image:2.595.313.558.292.464.2]Gaps are introduced in the sequence to get the best possible alignment, and in this manner it is very necessary to adopt a scheme for penalizing these gaps. The well-known approach is the affine gap penalty model where an initial penalty to start a gap is more than extending the gap an extra position [5].
Figure 5. Blosum62 Substitution Matrix [6].
II. METHODS OF SEQUENCE ALIGNMENT
Many algorithms for sequence alignment have been proposed for local, global and multiple sequence alignment. The different types are categorized into further algorithms as shown in Fig. 6 These algorithms come under two categories: optimal and heuristic alignment algorithms. Optimal alignment algorithms are slow but guaranteed to find best alignments. Heuristic algorithms are faster but less precise [7]. The alignment algorithms are discussed in following subsections.
A. Optimal alignment algorithm
For the pairwise sequence alignment, Dynamic Programming (DP) builds a two dimensional matrix, where two axes of the matrix are for the two sequences [8]. In DP all pairs of residues are exhaustively aligned, as per a scoring scheme for matches, gaps and mismatches.
[image:2.595.34.280.566.744.2]Figure 6. Different types of sequence alignments methods.
For optimal global sequence alignment Needleman– Wunsch algorithm is used, whereas the Smith-Waterman Algorithm produces the optimal local sequence alignment.
Dynamic Programming produce optimal alignment, but is complex and demands more computer resources for multiple sequence alignment. Therefore, to diminish this computational complexity, different heuristic methodologies have been created, but they don't ensure the global optimum.
B. Heuristic algorithms
These algorithms can give a solution to a problem; but they don't ensure finding the global optimum. BLAST [10]
and FASTA [11] are the most commonly utilized heuristic algorithms for pairwise alignment. These begin by identifying the list of seeds for the query that matched with the database sequence. Highly similar regions are referred to as a seed. After finding seeds, for each seed in the sequence extend seed alignment in both directions to get the desired pattern. BLAST and FASTA are best known for their execution in the large database search.
For multiple sequence alignment Clustal W, MUSCLE, T-Coffee package are the most commonly utilized heuristic algorithms.
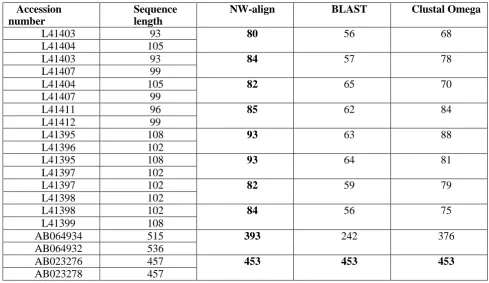
Table I. Comparative alignment results of DNA sequences on the basis of identities produces bt different tools.
Accession number
Sequence length
NW-align BLAST Clustal Omega
L41403 93 80 56 68
L41404 105
L41403 93 84 57 78
L41407 99
L41404 105 82 65 70
L41407 99
L41411 96 85 62 84
L41412 99
L41395 108 93 63 88
L41396 102
L41395 108 93 64 81
L41397 102
L41397 102 82 59 79
L41398 102
L41398 102 84 56 75
L41399 108
AB064934 515 393 242 376
AB064932 536
AB023276 457 453 453 453
[image:3.595.52.542.477.760.2]III. EXPERIMETNAL RESULT ANALYSIS
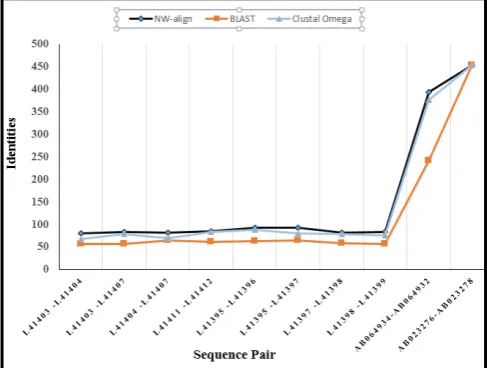
[image:4.595.40.284.239.423.2]In the experiment, we have compared the well-known sequence matching approaches: NW-Align, BLAST, Clustal Omega. NW-Align is based on dynamic programming, BLAST and Clustal Omega are huristic tools. The sequences with different lengths are chosen to observe the performance of different approaches for both the smaller and the larger length sequences. The identity values of these approaches have been tabulated in Table I. Sequences: L41403, L41404, L41407, L41411, L41412, L41395, L41396, L41397, L41398, L41399, AB064934, AB064932, AB023276, AB023278 are retrieved from NCBI: http://www.ncbi.nlm.nih.gov/ in the FASTA format.
Figure 9. Performance comparison of NW-Align, BLAST and Clustal Omega in terms of identities of DNA sequence pairs.
Bold face results in the table show the highest identity obtained by the respective method [12]. From Table 1, we can
see that the NW-Align performed better than other methods.
IV. SQUENCE ALIGNMENT TOOLS
Some of the important software tools for sequence alignment are listed below in Table 2. These are grouped into 3 categories: database search tools, Pairwise sequence alignment, Multiple sequence alignment [13]. They are also distinguished on the basis of the alignments they produce. Respective links of the tools are also provided.
V. CONCLUSIONS
This is a survey paper, aiming to provide a guide for researchers on the sequence alignment problem. Sequence alignment has been an extremely active research area and is one of the real issues as the biological databases size is continuously expanding. There has been an explosion of algorithms in this field. These algorithms can be classified as database search tools, pairwise and multiple sequence alignment tools. These algorithms can be further classified as optimal or heuristic and local or global. Optimal algorithms based on dynamic programming gives us the exact and optimal solutions, but is complex and demands more computer resources for multiple sequence alignment. As a solution, heuristics is considered as an option, they are much faster but they do not provide optimal solution. This is where, recently introduced nature motivated stochastic optimization methods gain importance. They can effectually take care of the complex multiple sequence alignment problem but need refinement to provide the better results.
TABLE II. SEQUENCE ALIGNMENT TOOLS.
Software type Tool Alignment type Link
A. Databas Search
BLAST Local https://blast.ncbi.nlm.nih.gov/Blast.cgi FASTA Local http://www.ebi.ac.uk/Tools/sss/fasta/
B. Pairwise sequence alignment
NW-align Global http://zhanglab.ccmb.med.umich.edu/NW-align/ Needle SemiGlobal http://www.ebi.ac.uk/Tools/psa/emboss_needle/ Water Local http://www.ebi.ac.uk/Tools/psa/emboss_water/
C. Multiple sequence alignment
Kalign Global http://www.ebi.ac.uk/Tools/msa/kalign/ Clustal
Omega
Local or global http://www.ebi.ac.uk/Tools/msa/clustalo/
MUSCLE Local or global http://www.ebi.ac.uk/Tools/msa/muscle/ T-Coffee Local or global http://www.ebi.ac.uk/Tools/msa/tcoffee/
VI. REFERENCES
[1] W. Haque, A. Aravind, and B. Reddy, “Pairwise sequence alignment algorithms: a survey”, In Proceedings of the conference on Information Science, Technology and
[2] M.O. Dayhoff, “Survey of new data and computer methods of
analysis” Atlas of protein sequence and structure, 1976..
[4] J. Cohen, “Bioinformatics—an introduction for computer scientists” ACM Computing Surveys (CSUR), vol. 36, no. 2, pp. 122-158, 2004.
[5] C. Gondro, and B. P. Kinghorn, “A simple genetic algorithm for multiple sequence alignment”, Genetics and Molecular Research, vol. 6, no. 4, pp. 964-982, 2007.
[6] J. Xiong, “Essential bioinformatics” Cambridge University Press, 2006.
[7] L. Hasan, and Z. Al-Ars, “An overview of hardware-based acceleration of biological sequence alignment”, INTECH Open Access Publisher, 2011.
[8] S. B. Needleman, and C. D. Wunsch, “A general method applicable to the search for similarities in the amino acid sequence of two proteins”, Journal of molecular biology, vol. 48, no. 3, pp. 443-453, 1970.
[9] E. S. Orabi, M. A. Assal, M. A. Azim, and Y. Kamal, “DNA fingerprint using smith waterman algorithm by grid computing”, 2014 9th IEEE International Conference on In Informatics and Systems (INFOS), December 2014
[10] S. F. Altschul, W. Gish, W. Miller, E. W. Myers, and D. J. Lipman, “Basic local alignment search tool”, Journal of molecular biology, vol. 215, no. 3, pp. 403-410, 1990.
[11] W. R. Pearson, “Rapid and sensitive sequence comparison with FASTP and FASTA”, Methods in enzymology, vol. 183, pp. 63-98, 1990.
[12] R. K. Jena, M. M. Aqel, P. Srivastava, and P.K. Mahanti, “Soft computing methodologies in bioinformatics”, European Journal of Scientific Research, vol. 26, no.2, pp. 189-203, 2009.
![Figure 5. Blosum62 Substitution Matrix [6].](https://thumb-us.123doks.com/thumbv2/123dok_us/688574.1076264/2.595.34.280.566.744/figure-blosum-substitution-matrix.webp)