Overlapping Roles of the Spindle Assembly and DNA Damage Checkpoints in
the Cell-Cycle Response to Altered Chromosomes in
Saccharomyces cerevisiae
Peter M. Garber and Jasper Rine
1Department of Molecular and Cell Biology, University of California, Berkeley, California 94720
Manuscript received July 31, 2001 Accepted for publication March 4, 2002
ABSTRACT
TheM A D2-dependent spindle checkpoint blocks anaphase until all chromosomes have achieved success-ful bipolar attachment to the mitotic spindle. The DNA damage and DNA replication checkpoints block anaphase in response to DNA lesions that may include single-stranded DNA and stalled replication forks. Many of the same conditions that activate the DNA damage and DNA replication checkpoints also activated the spindle checkpoint. Themad2⌬mutation partially relieved the arrest responses of cells to mutations affecting the replication proteins Mcm3p and Pol1p. Thus a previously unrecognized aspect of spindle checkpoint function may be to protect cells from defects in DNA replication. Furthermore, in cells lacking either the DNA damage or the DNA replication checkpoints, the spindle checkpoint contributed to the arrest responses of cells to the DNA-damaging agent methyl methanesulfonate, the replication inhibitor hydroxyurea, and mutations affecting Mcm2p and Orc2p. Thus the spindle checkpoint was sensitive to a wider range of chromosomal perturbations than previously recognized. Finally, the DNA replication checkpoint did not contribute to the arrests of cells in response to mutations affecting ORC, Mcm proteins, or DNA polymerase ␦. Thus the specificity of this checkpoint may be more limited than previously recognized.
T
HE ability to accurately transmit genetic material byFoianiet al. 2000). A second checkpoint, variously called the replication, S-phase, or S-M checkpoint, ar-to daughter cells is essential ar-to all life. Eukaryoticrests cells in which replication has been blocked by organisms have evolved mechanisms called checkpoints
deoxyribonucleotide depletion. This checkpoint over-that increase the fidelity of genetic transmission.
Check-laps with the DNA damage checkpoint in its require-points enhance fidelity by delaying cell-cycle
progres-ment forR A D53,MEC1, andDDC2, but unlike the DNA sion in cells with defects in chromosomes or in the
damage checkpoint, does not require R A D9, RAD17, machinery that segregates chromosomes. Cancer cells
R A D24, M EC3, or DDC1 (reviewed by Lowndes and display reduced fidelity of genetic transmission and
fre-Murguia2000). A third checkpoint, termed the spindle quently have mutations in checkpoint genes (Li and
assembly checkpoint, arrests cells in which replicated Benezra1996;Cahillet al.1998;Lengaueret al.1998).
chromatids fail to achieve bipolar spindle attachment. Thus checkpoint failure contributes to cancer. Since
This checkpoint requiresM A D1,M A D2,M A D3,BUB1, the defects that checkpoints respond to are likely to be
BUB3,NDC10, andM PS1for full function (reviewed by defects that destabilize the genome, identification of
Hoyt2001). those defects should increase our understanding of
ge-In this study, we quantify the roles of these three netic instability and also our understanding of how
checkpoints in the preanaphase arrests that occur in checkpoints prevent cancer.
cells that have lost the function of various essential repli-A variety of conditions that disrupt chromosomes
cation proteins. Initially, in an attempt to gain insight and/or chromosome segregation cause Saccharomyces
into the function of the eukaryotic DNA replication cerevisiaecells to arrest prior to anaphase through one or
initiator, the origin recognition complex (ORC), we more of three different checkpoints. These checkpoints
quantified the roles of these three checkpoints in the differ in the types of agents that elicit their response and
preanaphase arrests of cells that had lost ORC function. also in the genes that are required for their function. A
We were surprised to find that, although the DNA dam-checkpoint termed the DNA damage dam-checkpoint arrests
age and spindle assembly checkpoints contributed to cells that have been treated with DNA-damaging agents.
the arrests oforccells, the DNA replication checkpoint This checkpoint requiresRAD9,RAD17,RAD24,RAD53,
did not. To determine whether this pattern of check-DDC1,DDC2,MEC1, andMEC3for full function (reviewed
point responses was unique to orc mutants, we con-ducted similar analyses of cells harboring conditional mutations affecting Mcm proteins, DNA Pol␣, and DNA
1Corresponding author:Department of Molecular and Cell Biology,
Pol␦. We found that the spindle assembly and DNA
401 Barker Hall, University of California, Berkeley, CA 94720.
E-mail: [email protected] damage checkpoints jointly mediated arrest responses
RESULTS to a variety of replication mutations and that the spindle
assembly checkpoint was capable of mediating arrest To test the roles of the DNA damage, DNA replica-responses to a DNA-damaging agent and a DNA replica- tion, and spindle assembly checkpoints in the arrest
tion inhibitor. responses of cells with replication defects, budding yeast
strains harboring temperature-sensitive mutations in genes encoding one of five different replication proteins were studied. Three of these mutations affect proteins MATERIALS AND METHODS
involved in replication initiation:orc2-1(affecting ORC
Yeast strains: mcm2-1, mcm3-1, cdc2-1, and pol1-17 alleles
subunit 2);mcm2-1[affecting minichromosome mainte-from other strain backgrounds were backcrossed to W303 a
nance (MCM) protein 2]; andmcm3-1(affecting MCM minimum of four times. Theorc2-1strain was constructed by
protein 3) (Yan et al. 1991; Foss et al. 1993). These gene replacement ofORC2in W303. The resulting collection
of replication mutants was crossed to rad9⌬, rad24⌬, mad2, proteins are components of the preinitiation complex, and mec1⌬ checkpoint mutant strains isogenic to W303 to which assembles at replication origins, rendering them create the replication mutant strains used in this study (Table competent to initiate DNA replication. Intriguingly, mu-1). Themad1⌬::KanMXallele, derived from the Research
Ge-tations in each of these proteins cause cells to arrest netics (Birmingham, AL)MATadeletion collection, was
back-prior to anaphase with a genome that is either fully repli-crossed once to W303 before crossing to a W303-isogenic
cated or nearly so (Gibsonet al.1990;Yanet al.1991; rad9⌬rad24⌬strain to create JRY7309–JRY7320. Standard
ge-netic procedures were as described (Roseet al. 1989). Bellet al.1993;PflummandBotchan2001). Since it
Growth, synchronization, methyl methanesulfonate treat- was not clear why mutations affecting the preinitiation ment, hydroxyurea treatment, and 4ⴕ,6-diamidino-2-phenylin- complex should cause cells to arrest prior to anaphase, dole staining:YPD (rich medium) was used in all experiments.
knowledge of which checkpoints, if any, were responsi-Temperature-sensitive replication mutants were maintained
ble for this phenotype was a first step toward understand-at 23⬚. The restrictive temperature used in all
temperature-ing it. shift experiments was 37⬚. Strains lacking replication
muta-tions were grown at 25–28⬚.␣-Factor was used at 2.5–5g/ml The other two mutations affect proteins involved in to synchronizeMATacells in G1.␣-Factor-arrested cells were replication elongation. Thecdc2-1mutation affects DNA washed twice in prewarmed YPD before being released into
polymerase ␦, the major replicative DNA polymerase prewarmed YPD containing 5g/ml Pronase (Calbiochem
(Bouletet al.1989), and causes cells to arrest in mid-53702) protease. Methyl methanesulfonate (MMS, M-4016;
S-phase (BuddandCampbell 1993; P.Garberand J. Sigma, St. Louis) was added to YPD medium at 0.033%, except
as noted. Hydroxyurea (HU) was added to YPD medium at Rine, unpublished results). Thepol1-17mutation affects 200 mm. 4⬘,6-Diamidino-2-pheynylindole (DAPI) staining of DNA polymerase ␣ (Budd and Campbell 1987), re-fixed cells was as described (Roseet al.1989), and cell-cycle sponsible for priming DNA synthesis, and also causes arrest or progression was determined by calculating the
per-cells to arrest in mid-S-phase (Buddet al.1989). Whereas centage of large-budded uninucleate cells by fluorescence
mi-it was unclear whether unreplicated DNA would be pres-croscopy of⬎200 cells. All cultures were coded during scoring
ent during the late S/G2 arrests of the initiation mu-so that the scorer was blind to the genotype of the culture
being scored. tants, the mid-S arrest of these mutants ensured that
Viability in MMS: The number of colony-forming units significantly underreplicated chromosomes would be (CFU) per microliter in cultures of wild-type,mad2⌬,rad9⌬ present for potential detection by the various
check-rad24⌬, andmad2⌬rad9⌬rad24⌬cells was determined both
points. before and at various times after the addition of 0.033% MMS
In a current model of the DNA-responsive checkpoint by plating cells on non-MMS-containing medium and
count-ing colonies after 3 days of growth at 25⬚. At each time point pathways inS. cerevisiae,MEC1is essential to the check-after MMS addition, the viability of each culture was expressed point responses to both DNA damage and stalled repli-as the relation (CFU per microliter at that time point)/(CFU cation. In contrast,R A D9andR A D24are essential only per microliter before MMS addition). To determine the
sig-to the checkpoint response sig-to DNA damage and are nificance of the effect ofmad2⌬on the viability of therad9⌬
not required for the response to stalled replication (Fig-rad24⌬strains, the viabilities of both therad9⌬rad24⌬strains
ure 6a). In preliminary experiments with the replication and themad2⌬rad9⌬rad24⌬strains were normalized to the
mean viability of therad9⌬rad24⌬strains at that time point. mutants, we found that combined rad9⌬ and rad24⌬ This normalization permitted compiling the viability of these mutations relieved the cell-cycle arrests of all of the strains at all time points, which was then expressed as (viability
mutants other than pol1-17to the same degree as did of strains X)/(viability ofrad9⌬rad24⌬strains)⫾95%
confi-themec1⌬mutation. Therefore theR A D9- andR A D24 -dence limits.
independent,MEC1-dependent replication checkpoint
Growth rate:Six (wild-type,mad2⌬) or seven (rad9⌬rad24⌬,
mad2⌬rad9⌬rad24⌬) log-phase cultures of strains of the indi- pathway did not make a significant contribution to the cated genotypes were diluted to OD600ⵑ0.06 and grown for arrests of these mutants. Since use of themec1⌬mutation
3.5–5 hr at 25⬚. The ratios of the final ODs to the initial ODs prevents distinguishing between the contributions of were used to compute the doubling time of each culture,
the DNA replication and DNA damage checkpoints, we which was then normalized to the mean doubling time of the
used the combined rad9⌬ and rad24⌬ mutations and wild-type cultures. The means of these normalized doubling
The spindle checkpoint protein Mad2p arrested cells responses (Hoyt et al. 1991; Hardwick et al. 1999). However,rad9⌬rad24⌬mutations only partially relieved
in response to replication mutations:Activation of the
DNA damage or the spindle checkpoint causes budding the MMS-induced arrest. MMS-treated rad9⌬ rad24⌬ strains accumulated 45% large-budded uninucleate yeast cells to arrest with a large bud and an undivided
nucleus. This type of arrest can be quantified by fluo- cells compared to the 10–14% observed in the untreated strains (Figure 2a,rad9⌬rad24⌬vs.no treatment). The rescence microscopic determination of the percentage
of large-budded uninucleate cells in a population. After mad2⌬mutation significantly reduced this residual accu-mulation to ⵑ26% (Figure 2a,rad9⌬rad24⌬vs. mad2⌬ creating yeast strains that harbored the replication
mu-tations as well as mumu-tations in the DNA damage check- rad9⌬rad24⌬). Thus, Mad2p was able to arrest a portion of MMS-treated cells and did so when the DNA damage point (rad9⌬rad24⌬), the spindle checkpoint (mad2⌬),
or both, we incubated cultures of these strains at the checkpoint was not present.
Although Mad2p is not known to have a function restrictive temperature and then determined the
per-centage of large-budded uninucleate cells in each. outside of its role in the spindle checkpoint, we consid-ered the possibility that the Mad2p-dependent arrest In strains in which both the DNA damage and DNA
replication checkpoints were intact,mad2⌬significantly responses in our experiments reflected a spindle check-point-independent function of Mad2p. To test this possi-reduced the arrests ofmcm3-1andpol1-17strains (Figure
1; checkpoint⫹ vs. mad2⌬). Thus, the full arrest re- bility, we determined whether inactivation of a different component of the spindle checkpoint, Mad1p, would sponse to these mutations required Mad2p. The effect
of the mad2⌬ mutation on mcm2-1, cdc2-1, and orc2- also relieve cell-cycle arrest responses to DNA damage. Similarly to mad2⌬, the mad1⌬ mutation significantly 1 strains was highly variable; thus whether the arrest
responses to these mutations required Mad2p was not reduced the arrest response of rad9⌬ rad24⌬ cells to MMS (Figure 2b). Thus, two different spindle check-resolved by this experiment. However, Mad2p did
con-tribute to the residual arrests ofmcm2-1andorc2-1strains point genes each promoted cell-cycle arrest in response to DNA damage. The simplest interpretation of this lacking the DNA damage checkpoint (Figure 1;
com-pare the rad9⌬ rad24⌬ double mutant to the rad9⌬ finding is that the spindle checkpoint itself promotes cell-cycle arrest in response to DNA damage.
rad24⌬mad2⌬triple mutant). Furthermore, Mad2p, in
conjunction with Rad9p and Rad24p, was required for The cell-cycle arrest defect ofrad9⌬rad24⌬cells rela-tive tomad2⌬cells treated with MMS indicated that the the full arrest response to thecdc2-1mutation (Figure 1;
comparecdc2-1withcdc2-1 mad2⌬rad9⌬rad24⌬). Thus, spindle checkpoint was less efficient at mediating this response than was the DNA damage checkpoint. One Mad2p, and therefore presumably also the spindle
checkpoint, detectably responded to the altered chro- explanation for this difference could be that the DNA damage checkpoint recognizes most MMS-induced le-mosomes generated in each of the replication mutants
studied. These included mutations affecting both the sions whereas the spindle checkpoint recognizes only a subset of them. For example, only a subset of the MMS-initiation and elongation of replication and mutations
causing cells to arrest either in mid-S-phase or in late induced lesions might interfere with centromere func-tion and hence activate the spindle checkpoint. If this S-G2.The combined mad2⌬rad9⌬rad24⌬ mutations
eliminated the accumulation of large-budded uninucle- model were correct, then it should be possible to reduce the concentration of MMS to a level at which most or ate cells inmcm2-1,mcm3-1,orc2-1, and cdc2-1 cultures
held at the restrictive temperature (Figure 1; mad2⌬ all cells experience a lesion that activates the damage checkpoint, while only a subset of cells experience a rad9⌬rad24⌬vs.no treatment). Thus, in the absence of
these checkpoints, none of these replication mutations lesion that activates the spindle checkpoint. Therefore, reduced MMS concentrations were evaluated for their created a block to anaphase progression. Moreover,
al-though MEC1 was intact in these strains, they failed effects on the arrests ofmad2⌬andrad9⌬rad24⌬strains. A fourfold reduction in MMS concentration had no to arrest. Therefore, inactivation of these replication
proteins failed to detectably activate theMEC1-depen- detectable effect on the arrest of the mad2⌬ strains, presumably reflecting the ability of the DNA damage dent, R A D9 RAD24-independent replication
check-point. checkpoint to respond to low levels of MMS-induced
damage. In contrast, the lower MMS concentration re-The spindle checkpoint arrested cells in response to
methyl methanesulfonate and hydroxyurea: These re- duced the arrest of therad9⌬rad24⌬strains from 45 to
25% (Figure 2c). These data suggested that only a subset sults with replication mutants suggested that the spindle
checkpoint may be sensitive to a wide range of DNA pertur- of MMS-induced lesions could activate the spindle checkpoint.
bations. To explore this range further, the ability of
Mad2p to arrest cells treated with the DNA-damaging To explore further the spindle checkpoint’s ability to respond to DNA perturbations, the ability ofmad2⌬to agent MMS was tested. No effect ofmad2⌬was observed
on the arrests of strains with an intact DNA damage relieve the arrest response to an agent that stalls DNA replication was tested. HU stalls DNA replication by checkpoint (Figure 2a), consistent with previous reports
TABLE 1
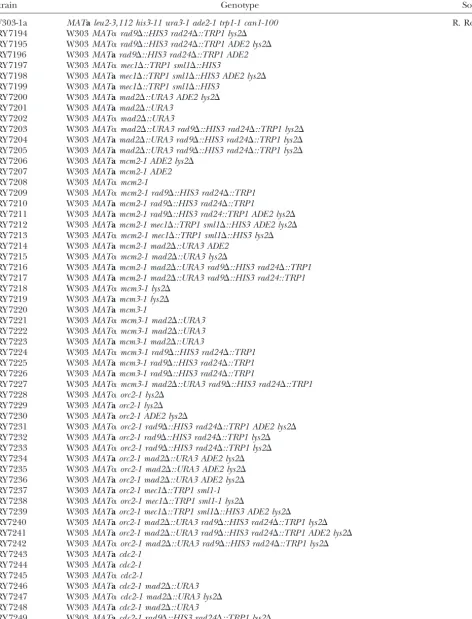
Yeast strains used in this study
Strain Genotype Sourcea
W303-1a MATaleu2-3,112 his3-11 ura3-1 ade2-1 trp1-1 can1-100 R. Rothstein JRY 7194 W303MAT␣rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬
JRY 7195 W303MAT␣rad9⌬::HIS3 rad24⌬::TRP1 ADE2 lys2⌬ JRY7196 W303MATarad9⌬::HIS3 rad24⌬::TRP1 ADE2
JRY 7197 W303MAT␣mec1⌬::TRP1 sml1⌬::HIS3
JRY 7198 W303MATamec1⌬::TRP1 sml1⌬::HIS3 ADE2 lys2⌬
JRY 7199 W303MATamec1⌬::TRP1 sml1⌬::HIS3
JRY 7200 W303MATamad2⌬::URA3 ADE2 lys2⌬
JRY 7201 W303MATamad2⌬::URA3
JRY 7202 W303MAT␣mad2⌬::URA3
JRY 7203 W303MAT␣mad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬ JRY 7204 W303MATamad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬
JRY 7205 W303MATamad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬
JRY 7206 W303MATamcm2-1 ADE2 lys2⌬
JRY 7207 W303MATamcm2-1 ADE2
JRY 7208 W303MAT␣mcm2-1
JRY 7209 W303MAT␣mcm2-1 rad9⌬::HIS3 rad24⌬::TRP1 JRY 7210 W303MATamcm2-1 rad9⌬::HIS3 rad24⌬::TRP1
JRY 7211 W303MATamcm2-1 rad9⌬::HIS3 rad24::TRP1 ADE2 lys2⌬
JRY 7212 W303MATamcm2-1 mec1⌬::TRP1 sml1⌬::HIS3 ADE2 lys2⌬
JRY 7213 W303MAT␣mcm2-1 mec1⌬::TRP1 sml1⌬::HIS3 lys2⌬ JRY 7214 W303MATamcm2-1 mad2⌬::URA3 ADE2
JRY 7215 W303MAT␣mcm2-1 mad2⌬::URA3 lys2⌬
JRY 7216 W303MATamcm2-1 mad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1
JRY 7217 W303MATamcm2-1 mad2⌬::URA3 rad9⌬::HIS3 rad24::TRP1
JRY 7218 W303MAT␣mcm3-1 lys2⌬ JRY 7219 W303MATamcm3-1 lys2⌬
JRY 7220 W303MATamcm3-1
JRY 7221 W303MAT␣mcm3-1 mad2⌬::URA3 JRY 7222 W303MAT␣mcm3-1 mad2⌬::URA3 JRY 7223 W303MATamcm3-1 mad2⌬::URA3
JRY 7224 W303MAT␣mcm3-1 rad9⌬::HIS3 rad24⌬::TRP1 JRY 7225 W303MATamcm3-1 rad9⌬::HIS3 rad24⌬::TRP1
JRY 7226 W303MATamcm3-1 rad9⌬::HIS3 rad24⌬::TRP1
JRY 7227 W303MAT␣mcm3-1 mad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1 JRY 7228 W303MAT␣orc2-1 lys2⌬
JRY 7229 W303MATaorc2-1 lys2⌬
JRY 7230 W303MATaorc2-1 ADE2 lys2⌬
JRY 7231 W303MAT␣orc2-1 rad9⌬::HIS3 rad24⌬::TRP1 ADE2 lys2⌬ JRY 7232 W303MATaorc2-1 rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬
JRY 7233 W303MAT␣orc2-1 rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬ JRY 7234 W303MATaorc2-1 mad2⌬::URA3 ADE2 lys2⌬
JRY 7235 W303MAT␣orc2-1 mad2⌬::URA3 ADE2 lys2⌬ JRY 7236 W303MATaorc2-1 mad2⌬::URA3 ADE2 lys2⌬
JRY 7237 W303MATaorc2-1 mec1⌬::TRP1 sml1-1
JRY 7238 W303MAT␣orc2-1 mec1⌬::TRP1 sml1-1 lys2⌬
JRY 7239 W303MATaorc2-1 mec1⌬::TRP1 sml1⌬::HIS3 ADE2 lys2⌬
JRY7240 W303MATaorc2-1 mad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬
JRY7241 W303MATaorc2-1 mad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1 ADE2 lys2⌬
JRY7242 W303MAT␣orc2-1 mad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬ JRY 7243 W303MATacdc2-1
JRY 7244 W303MATacdc2-1
JRY 7245 W303MAT␣cdc2-1
JRY 7246 W303MATacdc2-1 mad2⌬::URA3
JRY 7247 W303MAT␣cdc2-1 mad2⌬::URA3 lys2⌬ JRY 7248 W303MATacdc2-1 mad2⌬::URA3
JRY 7249 W303MATacdc2-1 rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬
TABLE 1
(Continued)
Strain Genotype Sourcea
JRY 7250 W303MAT␣cdc2-1 rad9⌬::HIS3 rad24⌬::TRP1 JRY 7251 W303MATacdc2-1 rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬
JRY 7252 W303MATacdc2-1 mad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1
JRY 7253 W303MAT␣cdc2-1 mad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1 JRY 7254 W303MATacdc2-1 mad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬
JRY 7256 W303MATapol1-17 lys2⌬
JRY 7257 W303MATapol1-17 lys2⌬
JRY 7258 W303MATapol1-17 mec1⌬::TRP1 sml1⌬::HIS3
JRY 7259 W303MAT␣pol1-17 mec1⌬::TRP1 sml1⌬::HIS3 JRY 7260 W303MATapol1-17 mec1⌬::TRP1 sml1⌬::HIS3
JRY 7261 W303MATapol1-17 mad2⌬::URA3
JRY 7262 W303MAT␣pol1-17 mad2⌬::URA3 JRY 7263 W303MATapol1-17 mad2⌬::URA3 lys2⌬
JRY 7264 W303MAT␣pol1-17 rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬ JRY 7265 W303MAT␣pol1-17 rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬ JRY 7267 W303MAT␣pol1-17 mad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1 JRY 7268 W303MATapol1-17 mad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1 lys2⌬
JRY 7269 W303MAT␣pol1-17 mad2⌬::URA3 rad9⌬::HIS3 rad24⌬::TRP1 JRY 7270 W303MATamcm2-1 rad24⌬::TRP1 lys2⌬
JRY 7271 W303MATamcm2-1 mad2⌬::URA3 rad24⌬::TRP1 ADE2
JRY 7272 W303MAT␣rad9⌬::HIS3 rad24⌬::TRP1 ADE2 lys2⌬ JRY 7274 W303MATamec1⌬::TRP1 sml1-1
JRY 7275 W303MATamec1⌬::TRP1 sml1⌬::HIS3 mad2⌬::URA3
JRY 7309 MATaleu2-3,112 his3-11 ura3-1 ade2-1 trp1-1
JRY 7310 MAT␣leu2-3,112 his3-11 ura3-1 ade2-1 trp1-1lys2⌬ JRY 7311 MATaleu2-3,112 his3-11 ura3-1 ade2-1 trp1-1
JRY 7312 MAT␣leu2-3,112 his3-11 ura3-1 ade2-1 trp1-1 rad9⌬::HIS3 rad24⌬::TRP1 JRY 7313 MATaleu2-3,112 his3-11 ura3-1 ade2-1 trp1-1 rad9⌬::HIS3 rad24⌬::TRP1
JRY 7314 MATaleu2-3,112 his3-11 ura3-1 ade2-1 trp1-1 rad9⌬::HIS3 rad24⌬::TRP1
JRY 7315 MATaleu2-3,112 his3-11 ura3-1 ade2-1 trp1-1 mad1⌬::KanMX
JRY 7316 MAT␣leu2-3,112 his3-11 ura3-1 ade2-1 trp1-1 mad1⌬::KanMX JRY 7317 MATaleu2-3,112 his3-11 ura3-1 ade2-1 trp1-1 mad1⌬::KanMX
JRY 7318 MATaleu2-3,112 his3-11 ura3-1 ade2-1 trp1-1 rad9⌬::HIS3 rad24⌬::TRP1 mad1⌬::KanMX
JRY 7319 MAT␣leu2-3,112 his3-11 ura3-1 ade2-1 trp1-1 rad9⌬::HIS3 rad24⌬::TRP1 mad1⌬::KanMX JRY 7320 MATaleu2-3,112 his3-11 ura3-1 ade2-1 trp1-1 rad9⌬::HIS3 rad24⌬::TRP1 mad1⌬::KanMX
All strains are congenic with W303-1a, with additional mutations as noted.
aExcept as noted, all strains were produced for this study.
cells of deoxyribonucleotides. This condition activates damage, this result indicated that incompletely repli-cated chromosomes per se may activate the spindle the replication checkpoint, which is independent of
R A D9 and R A D24 but requires MEC1. Therefore we checkpoint.
The checkpoints acted in the first cell cycle following tested the ability ofmad2⌬to relieve arrest in
hydroxy-urea-treated mec1⌬ cells, which lack the replication damage:In the preceding experiments, cell-cycle arrest was quantified in cultures that were growing asynchro-checkpoint. After 3.5 hr in 200 mmHU,ⵑ80% of
wild-type cells or cells lacking the DNA damage checkpoint nously at the time of insult. When cells fail to accumulate at the arrest point in such an experiment, they may do arrested (Figure 2d). Themec1⌬cells arrested less well,
yet still exhibited more cell-cycle arrest than has been so either by passing through the arrest point (a checkpoint defect) or by failing to ever arrive at the arrest point reported by others (Figure 2d, wild typevs. mec1⌬;
Wein-ertet al.1994;Desanyet al.1998). At later time points (a viability defect). Experiments on synchronized cell populations allowed us to distinguish between these pos-mec1⌬relieved a larger portion of the HU-induced arrest
(Figure 5 and data not shown), possibly explaining this sibilities. This question was relevant to the mechanism of spindle-checkpoint-mediated arrest. Mitosis in the discrepancy. mad2⌬ significantly reduced the residual
arrest observed in HU-treatedmec1⌬ cells (Figure 2d, presence of damaged or partially replicated chromo-somes can lead to aneuploidy and chromosome break-mec1⌬vs. mec1⌬mad2⌬). Since HU treatment stalls
Figure1.—M A D2-mediated arrest responses to a variety of replication mutations. Cultures of strains of the indicated genotypes were incubated at the restrictive temperature of 37⬚ for 3.5 hr and then harvested, fixed, and stained with DAPI. The percentage of large-budded uninucleate cells in each culture was then determined by fluores-cence microscopic examination of ⬎200 cells. Each bar represents the mean of 3–11 such trials, and the error bars represent the 95% confidence limits of that mean. Excepting mcm3-1 rad9⌬
rad24⌬, each genotype was represented by a mini-mum of two, and usually three or more, indepen-dently derived strains. “No treatment” refers to control strains lacking replication mutations but containing the indicated checkpoint mutations.
activate the spindle checkpoint inS. cerevisiae(Wellsand age checkpoint arrested this proportion of cells in the first cycle following inactivation of Mcm2p. Similarly, Murray1996), it was possible that the spindle checkpoint
responses to MMS, HU, or replication mutations re- 45% of the mcm2-1 rad24⌬ cells accumulated at the arrest point, indicating that the spindle checkpoint ar-quired a prior mitosis in the presence of these insults.
To test these ideas, we synchronized cells in G1 with rested this proportion of cells in the first cycle following inactivation of Mcm2p. Thus, both DNA damage
check-␣-factor prior to the time of insult and then quantified
the number of cells both arriving at and passing through point- and spindle checkpoint-mediated arrest occurred in the first cycle after inactivation of Mcm2p.
the arrest point.
In the first synchronized-cell experiment, themcm2-1 To assess checkpoint responses in the first cell cycle after exogenously induced DNA damage, the experi-mutation was used to activate the checkpoints. R A D9
was left intact in the strains used in this experiment ment was repeated using MMS rather than mcm2-1 to activate the checkpoints. Wild-type,rad9⌬rad24⌬,mad2⌬, sincerad9⌬in combination with other mutations caused
low viability that prevented synchronization. Themcm2-1, andmad2⌬rad9⌬rad24⌬cells were synchronized in G1 and then released into medium containing MMS. In mcm2-1 mad2⌬,mcm2-1 rad24⌬, andmcm2-1 mad2⌬rad24⌬
strains were synchronized in G1 at the permissive tem- this experiment, bud emergence and growth occurred with similar kinetics in all the strains (Figure 4, a and perature and then released from the G1 block into
re-strictive-temperature medium. Under these conditions, b). In cells with both checkpoints intact (WT in Figure 4) and in cells with just the DNA damage checkpoint bud emergence and bud growth occurred with similar
kinetics in themcm2-1 and mcm2-1 mad2⌬ strains, but intact (mad2⌬),⬎80% of the population arrested (Fig-ure 4c). In cells with only the spindle checkpoint intact were slightly slower and/or less synchronous in themcm2-1
rad24⌬andmcm2-1 rad24⌬ mad2⌬ strains (Figure 3, a (rad9⌬rad24⌬), 55% of the population arrested (Figure 4c). However, in cells with neither checkpoint intact and b). By 100 min postrelease, 80% of themcm2-1cells
with both checkpoints intact accumulated at the large- (mad2⌬ rad9⌬ rad24⌬), only 14% of the population arrested (Figure 4c). Cell-cycle progression in the triple budded uninucleate stage (Figure 3c,mcm2-1). By
con-trast, only 12% of mcm2-1 cells lacking both the DNA mutant cells was confirmed by subsequent increases in unbudded cells, small-budded cells, and large-budded damage and spindle checkpoints accumulated at this
stage (Figure 3c, mcm2-1 mad2⌬ rad24⌬). Subsequent binucleate cells (Figure 4, a, b, and d). Thus both DNA damage checkpoint activation and spindle checkpoint increases in the proportions of binucleate and
unbud-ded cells in this strain demonstrated that these cells activation occurred in the first cycle after treatment with MMS.
were passing through the arrest point rather than failing
to arrive at it (Figure 3d,mcm2-1 mad2⌬rad24⌬; Figure To explore further the role ofM A D2in the arrest of mec1⌬cells in HU, we monitored the cell-cycle progres-3a,mcm2-1 mad2⌬rad24⌬). Thus, the preanaphase
ar-rest of mcm2-1cells was due solely to the responses of sion of wild-type,mec1⌬, andmec1⌬mad2⌬cells released from the ␣-factor block into HU-containing medium. the DNA damage and spindle assembly checkpoints to
themcm2-1mutation. In this experiment, all three strains behaved similarly up until 120 min after release into HU-containing medium, Furthermore, 55% of themcm2-1 mad2⌬cells
Figure 2.—The spindle-checkpoint-arrested cells lacking the DNA damage or DNA replication checkpoints in response to MMS and HU. The percentage of large-budded uninucleate cells in each culture was determined as in Figure 1. (a) Cultures of strains with the indicated checkpoint mutations were treated with 0.033% MMS for 3.5 hr prior to fixation. From 7 to 11 cultures of each genotype were tested, and the mean and 95% confidence limits of that mean are shown. (b) Experiments were performed as in a. Three strains of each genotype were tested either two (mad1⌬) or three (wt,rad9⌬rad24⌬, mad1⌬rad9⌬
rad24⌬) times each, and the mean and 95% confidence limits are shown. (c) Three cultures each of amad2⌬strain and arad9⌬
rad24⌬strain were incubated with either 0.033% MMS (shaded) or 0.008% MMS (stippled) for 3.5 hr. Means and 95% confidence limits are shown. (d) Three cultures each of wild-type,mec1⌬, andmec1⌬mad2⌬strains were incubated with 200 mmhydroxyurea for 3.5 hr prior to analysis. Means⫾95% confidence limits are shown.
cells. Thus, the initial cell-cycle arrest response to HU tants:Cell-cycle delay in the presence of DNA damage often preserves cell viability, and indeed many check-occurred as efficiently in themec1⌬mad2⌬strain as in
the mec1⌬ strain (Figure 5c). However, by 150 min, point mutants have been identified by their sensitivity to DNA-damaging agents. Therefore we tested whether the percentage of large-budded uninucleate cells in the
wild-type strain continued to increase, whereas this per- themad2⌬mutation affected survival of MMS treatment. In tests for growth on solid MMS-containing medium, centage remained constant in themec1⌬strain, and in
the mec1⌬ mad2⌬ strain it declined. The rise in the themad2⌬mutation did not significantly affect the sur-vival of either wild-type cells orrad9⌬rad24⌬cells (data percentage of binucleate cells during this time indicated
that the loss of uninucleate cells was due to nuclear not shown). Thus spindle checkpoint function did not enhance survival of cells during chronic exposure to division (Figure 5d). Thus the spindle checkpoint was
able to block nuclear division in a portion of HU-treated MMS. However, to test whether spindle checkpoint func-tion could rescue cells from acute exposure to MMS, mec1⌬cells. Since the initial arrest response to HU
oc-curred as efficiently in themec1⌬mad2⌬strain as it did we treated wild-type, mad2⌬, rad9 rad24⌬, and mad2⌬ rad9⌬rad24⌬cells in liquid culture with MMS, removing in the mec1⌬ strain, these results suggested a role for
M A D2in maintaining the anaphase block in HU-treated cells at various times to test viability. This analysis re-vealed that the viability of the mad2⌬ rad9⌬ rad24⌬ cells rather than in establishing the block.
The spindle checkpoint contributed to the growth strains (39⫾12% relative torad9⌬rad24⌬viability) was
significantly lower than that of therad9⌬rad24⌬strains
Figure
4.—The
spindle-checkpoint-arrested
cells
in
the
first
cell
cycle
after
MMS
exposure.
Cultures
of
(
䉬
)
w
ild-type
(
JRY2334),
(
䉱
)
mad2
⌬
(
JRY7201),
(
䊏
)
rad9
⌬
rad24
⌬
(
JRY7196),
and
(
䊉
)
mad2
⌬
rad9
⌬
rad24
⌬
(
JRY7204)
cells
were
arrested
in
G1
with
␣
-factor
and
then
released
into
␣
-factor-free
medium
containing
0
.033%
MMS.
At
the
indicated
time
points,
aliquots
w
ere
removed
and
fixed
for
DAPI
staining.
At
each
time
p
oint,
⬎
200
cells
of
each
strain
were
scored
as
unbudded
(a),
small-budded
(b),
large-budded
uninucleate
(c),
large-budded
w
ith
dividing
nucleus
(not
shown),
o
r
large-budded
b
inucleate
Figure
5.—The
spindle-checkpoint-arrested
cells
in
the
first
cell
cycle
after
HU
treatment.
Cultures
of
(
䉱
)
w
ild-type
(
JRY2334),
(
䉬
)
mec1
⌬
(
JRY7274),
a
nd
(
䊏
)
mec1
⌬
mad2
⌬
(
JRY7275)
cells
were
arrested
in
␣
-factor
a
nd
then
released
into
␣
-factor-free
medium
containing
2
00
m
m
HU.
At
the
indicated
time
points
aliquots
w
ere
removed
and
fixed
for
DAPI
staining.
At
each
time
point,
⬎
200
cells
of
each
strain
were
scored
as
unbudded
(a),
small-budded
(b),
large-budded
uninucleate
(c),
large-budded
with
dividing
nucleus
(not
shown),
o
r
large-budded
binucleate
(100⫾ 12%) at all times after MMS treatment. Thus, tions caused cells to arrest in early S-phase (the DNA polymerase mutations, hydroxyurea treatment, and MMS spindle checkpoint function did contribute to the
viabil-ity of rad9⌬ rad24⌬ cells during short-term exposure treatment), mono-oriented kinetochores on unrepli-cated centromeres were likely sources of spindle check-to MMS.
Relative to wild-type yeast strains, the mad2⌬ rad9⌬ point activation in these cells. However, theorcandmcm mutations caused cells to arrest in late S-phase or G2. rad24⌬strains formed small colonies. To quantify this
effect, we determined the doubling times of wild-type, Since unreplicated centromeres were less likely to be present in these cells, spindle checkpoint activation in mad2⌬,rad9⌬rad24⌬, andmad2⌬rad9⌬rad24⌬strains.
The growth of themad2⌬ strain was indistinguishable these mutants may signal defects in sister chromatid cohesion or may reflect heretofore unsuspected roles from wild type (99⫾3.6% of wild-type doubling time).
Therad9⌬rad24⌬strain grew more slowly (107⫾3.3% of ORC and MCM proteins in promoting the bipolar attachment of chromosomes to the mitotic spindle. of wild-type doubling time), and themad2⌬ mutation
enhanced this defect (117⫾3.4% of wild-type doubling The lesions recognized by the DNA damage and DNA replication checkpoints have not been determined as time). Since the cell-cycle data presented above
indi-cated that the checkpoints that these genes control re- precisely as that of the spindle checkpoint. It has been proposed that singstranded DNA (ssDNA) is the le-spond to aberrant replication, the slower doubling times
of these strains likely reflected a requirement for check- sion recognized by the DNA damage checkpoint. Corre-lations between the presence of ssDNA and DNA dam-point response to aberrant replication events during
normal cell divisions. age checkpoint activation support this model (Lydall
and Weinert 1995; Lee et al. 1998). However, since ssDNA is an expected intermediate in the processing of DISCUSSION
most types of DNA damage, this correlation is compati-ble with other models as well. Therefore, we interpret We investigated the relative contributions of the DNA
damage, DNA replication, and spindle assembly check- the DNA damage checkpoint activation in our experi-ments to signal the presence of some DNA lesion that points to the preanaphase arrest responses that occur
inS. cerevisiaecells exposed to a variety of chromosome- may be an intermediate in DNA damage processing that may or may not be ssDNA.
perturbing conditions. These conditions included
mu-tations affecting the origin recognition complex, Mcm The molecular defect responsible for activation of the MEC1-dependent,R A D9-independent DNA replication proteins, DNA polymerase␣, and DNA polymerase ␦,
as well as nucleotide depletion and exposure to a DNA- checkpoint is widely thought to be ongoing or stalled replication forks (reviewed byLowndesandMurguia damaging agent. Several findings arose from this work.
First, the spindle checkpoint was able to contribute to 2000). However, our results, in conjunction with data from other labs, suggest that this view may need refinement. the arrest responses to all of the conditions tested.
Sec-ond, spindle checkpoint function was essential for cells Specifically, the preanaphase arrest responses to a vari-ety of conditions in which replication is stalled or ongo-to achieve a full arrest response ongo-to mutations affecting
Mcm3p and Pol1p. Third, the R A D9-independent, ing requireR A D9; thus, these conditions all fail to sig-nificantly activate the R A D9-independent replication MEC1-dependent replication checkpoint made a
detect-able contribution to the arrests ofpol1-17mutants and checkpoint. For example, the preanaphase arrest re-sponses to thecdc2-1andcdc2-2mutations, which inacti-HU-treated cells, but not to any of the other conditions
tested. vate DNA Pol␦, requireR A D9(WeinertandHartwell
1993; P.Garberand J.Rine, unpublished results). Simi-Identification of the checkpoints that become
acti-vated under a certain condition should offer insight into larly, although two-dimensional gel analyses indicate that arrestedorc2-1,orc5-1, andmcm2-1mutant cells con-the molecular defects associated with that condition.
In the case of the spindle checkpoint, the activating tain replication forks, these mutants requireR A D9for preanaphase arrest (Lianget al. 1995;Leiet al. 1997). molecular defect has been well characterized. A large
body of evidence indicates that kinetochores not under Furthermore, yeast cells harboring an origin-deficient artificial chromosome requireR A D9to stably maintain tension from the mitotic spindle cause this checkpoint
to become activated (Riederet al. 1995;Chenet al.1996; the chromosome (van Brabantet al. 2001). Thus, nei-ther the stalled forks in orcand mcmmutants nor the Li andNicklas 1997). Some of this evidence derives
from studies ofS. cerevisiae, in which unreplicated chro- ongoing replication forks of the artificial chromosome significantly activate the R A D9-independent, MEC1 -mosomes and mosomes with defects in sister
chro-matid cohesion both activate the spindle checkpoint dependent replication checkpoint.
In our experiments and in those of others, the replica-(Castanoet al.1996;Skibbenset al.1999;Hannahet
al.2001;Mayeret al.2001;SternandMurray2001). tion checkpoint contributed to the arrests of deoxyribo-nucleotide-depleted cells and cells lacking Pol␣ DNA Similarly, incomplete DNA replication in Drosophila
condi-Figure6.—(a) A model of checkpoint pathways involved in responding to stalled replication and DNA damage. TheMEC1
pathway is the cell’s most sensitive DNA-responsive checkpoint. The response to DNA-damaging agents such as MMS and ionizing radiation requiresR A D9, theR A D24epistasis group,MEC1, and additional downstream protein kinases. In contrast, the response to the replication inhibitor hydroxyurea requiresMEC1, but does not requireR A D9or theR A D24epistasis group. The response to mitotic spindle disruptors requiresM A D2 and occurs independently ofMEC1. Both the MEC1-dependent checkpoint and the M A D2-dependent checkpoint block anaphase by stabilizing the anaphase inhibitor Pds1p. R A D9and theR A D24 group activate this pathway in response to DNA damage and DNA structures resulting from stalled or aberrant DNA replication. (b) Enhancements to the model derived from results presented here. The MEC1-dependent pathway’s requirement forR A D9and theR A D24group was abrogated only inpol␣mutants and hydroxyurea-treated cells. The hydroxyurea effect may be mediated by the impact of lowered deoxynucleotide pools on Pol␣. TheM A D2-dependent spindle checkpoint was less sensitive than the
MEC1-dependent pathway to most chromosome perturbations, but was equally or more sensitive than the MEC1-dependent pathway to certain aberrant chromosome structures such as those found inpol1-17andmcm3-1mutants.
1994; P.Garberand J.Rine,unpublished results; Fig- checkpoint in S. cerevisiae. A model that takes into ac-count this more limited role of the replication check-ures 2 and 3c). Since stalled replication structcheck-ures are
likely to be present in orc, mcm, and pol␦ mutants as point in responding to replication problems is pre-sented in Figure 6b.
well as in pol␣mutants and HU-treated cells, we propose
that the replication checkpoint, rather than recognizing Although the spindle checkpoint was capable of ar-resting cells in response to all of the chromosome-stalled replication structures per se, recognizes a DNA
lesion that is specific to cells lacking Pol␣DNA polymer- perturbing conditions we employed, the responses to mcm2-1,orc2-1, HU, and MMS did not require spindle ase activity and cells lacking deoxyribonucleotides. When
considering what lesion might be common to these two checkpoint function as long as the MEC1-dependent checkpoints were intact. Thus theMEC1-dependent check-conditions, we note that pol␣mutants and HU-treated
cells are each compromised for Pol␣DNA polymerase points responded readily to these conditions and medi-ated a maximal arrest response to them whether or not activity. Thus, one possibility is that the replication
check-point recognizes an intermediate in DNA replication the spindle checkpoint was present. By contrast, the MEC1-dependent checkpoints responded less readily to that persists only when Pol␣’s DNA polymerase is not
active. Studies inXenopus laevisextracts indicate that RNA themcm3-1andpol1-17mutations, and under these con-ditions the spindle checkpoint contributed to arrest primers activate the replication checkpoint. Hence, we
tions in the yeast DNA polymerase I gene. Proc. Natl. Acad. Sci.
to which they activate each of these checkpoints. On
USA84:2838–2842.
one end of the spectrum lie chromosome perturbations Budd, M. E., and J. L.Campbell, 1993 DNA polymerases␦andεare
required for chromosomal replication inSaccharomyces cerevisiae.
such as the presence of multiple linear
minichromo-Mol. Cell. Biol.13:496–505.
somes, which activate the spindle checkpoint without
Budd, M. E., K. D.Wittrup, J. E.Baileyand J. L.Campbell, 1989
apparently activating the DNA damage checkpoint (Wells DNA polymerase I is required for premeiotic DNA replication
and sporulation but not for X-ray repair inSaccharomyces cerevisiae.
andMurray1996). On the other end of the spectrum
Mol. Cell. Biol.9:365–376.
lie chromosome perturbations such as low levels of MMS
Cahill, D. P., C.Lengauer, J.Yu, G. J.Riggins, J. K.Willsonet al.,
and thecdc13-1mutation, which activate the DNA dam- 1998 Mutations of mitotic checkpoint genes in human cancers.
Nature392:300–303.
age checkpoint while causing little or no spindle
check-Castano, R. B., P. M. Brzoska, B. U. Sadoff, H. ChenandM. F.
point activation (Hardwicket al. 1999; P.Garberand
Christman, 1996 Mitotic chromosome condensation in the
J.Rine,unpublished results; Figure 2). Themcm3-1and rDNA requiresTRF4and DNA topoisomerase I inSaccharomyces
cerevisiae.Genes Dev.10:2564–2576.
pol1-17mutations fall between these two extremes. The
Chen, R.-H., J. C.Waters, E. D.Salmonand A. W.Murray, 1996
ability to thus classify the checkpoint responses to a
Association of spindle assembly checkpoint component XMAD2
particular mutation may lend insight into the underly- with unattached kinetochores. Science274:242–246.
Desany, B. A., A. A. Alcasabas, J. B. BachantandS. J. Elledge,
ing molecular phenotype of the mutants.
1998 Recovery from DNA replicational stress is the essential
The majority of cancers display a chromosome
insta-function of the S-phase checkpoint pathway. Genes Dev.12:2956–
bility phenotype (Lengauer et al. 1998). While some 2970.
Foiani, M., A. Pellicioli, M. Lopes, C. Lucca, M. Ferrariet al.,
cancer cells are defective for spindle checkpoint
func-2000 DNA damage checkpoints and DNA replication controls
tion and/or the expression of spindle checkpoint genes
inSaccharomyces cerevisiae.Mutat. Res.451:187–196.
(LiandBenezra1996;Cahillet al.1998), the molecu- Foss, M., F. J. Mcnally, P. LaurensonandJ. Rine, 1993 Origin
recognition complex (ORC) in transcriptional silencing and DNA
lar basis for chromosome instability in most cancers
replication inS. cerevisiae.Science262:1838–1844.
remains unknown (Lengaueret al. 1998). Therefore,
Garner, M., S. van KreeveldandT. T. Su, 2001 mei-41andbub1 discovery of the lesions responsible for chromosome block mitosis at two distinct steps in response to incomplete DNA
replication inDrosophilaembryos. Curr. Biol.11:1595–1599.
instability remains a critical step in understanding
tu-Gibson, S. I., R. T. SuroskyandB. Tye, 1990 The phenotype of
morigenesis. The ability of a variety of replication
per-the minichromosome maintenance mutantmcm3is characteristic
turbations to activate the spindle checkpoint suggests of mutants defective in DNA replication. Mol. Cell. Biol. 10:
5707–5720.
that the proper attachment of chromosomes to the
mi-Hannah, J. S., E. S. Kroll, V. LundbladandF. A. Spencer, 2001
totic spindle can be disrupted by defects in replication.
Saccharomyces cerevisiae CTF18 andCTF4are required for sister
If so, then replication defects could contribute directly chromatid cohesion. Mol. Cell. Biol.21:3144–3158.
Hardwick, K. G., R. Li, C. Mistrot, R-H. Chen, P. Dannet al.,
to the segregation defects underlying much of the
chro-1999 Lesions in many different spindle components activate
mosome instability in cancer cells. Furthermore, the
the spindle checkpoint in budding yeast. Genetics152:509–518.
response of the spindle checkpoint to the same types Hoyt, M. A., 2001 A new view of the spindle checkpoint. J. Cell
Biol.154:909–912.
of perturbations as the DNA damage checkpoint
sug-Hoyt, M. A., L. TotisandB. T. Roberts, 1991 S. cerevisiaegenes
gests that mutations affecting these two pathways would
required for cell-cycle arrest in response to loss of microtubule
synergistically contribute to genome instability. Hence, function. Cell66:507–517.
Lee, S. E., J. K. Moore, A. Holmes, K. Umezu, R. D. Kolodneret al.,
tumor cells with spindle checkpoint mutations may be
1998 Saccharomyces Ku70, Mre11/Rad50, and RPA proteins
more sensitive to chemotherapeutic agents that damage
regulate adaptation to G2/M arrest after DNA damage. Cell94:
DNA than are cells with functional spindle checkpoints. 399–409.
Lei, M., Y. Kawasaki, M. R. Young, M. Kihara, A. Suginoet al., 1997 We thank Peter Burgers, Judith Campbell, Andrew Murray, Rodney
Mcm2 is a target of regulation by Cdc7-Dbf4 during the initiation Rothstein, Bik Tye, and Ted Weinert for yeast strains; Paul Kaufman
of DNA synthesis. Genes Dev.11:3365–3374.
and Judith Sharp for helpful discussions and protocols; Christopher Lengauer, C., K. W.Kinzlerand B.Vogelstein, 1998 Genetic Beh, Orna Cohen-Fix, Alexa Franco, Lena Hwang, Paul Kaufman, instabilities in human cancers. Nature396:643–649.
Jim Keck, Ann Kirchmaier, Judith Sharp, and Jeremy Thorner for Li, X., andR. B. Nicklas, 1997 Tension-sensitive kinetochore phos-comments on this manuscript; and anonymous reviewers for sugges- phorylation and the chromosome distribution checkpoint in
pray-ing mantid spermatocytes. J. Cell Sci.110:537–545. tions that improved both the substance and presentation of the
manu-Li, Y., and R. Benezra, 1996 Identification of a human mitotic script. This work was supported by a grant from the National Institutes
checkpoint gene: hsMad2. Science274:246–248. of Health (GM-31105) and by core support from a National Institute
Liang, C., M. WeinreichandB. Stillman, 1995 ORC and Cdc6p of Environmental Health Sciences Mutagenesis Center grant (P30
interact and determine the frequency of initiation of DNA replica-ES01896).
tion in the genome. Cell81:667–676.
Lowndes, N. F., andJ. R. Murguia, 2000 Sensing and responding to DNA damage. Curr. Opin. Genet. Dev.10:17–25.
Lydall, D., andT. Weinert, 1995 Yeast checkpoint genes in DNA
LITERATURE CITED
damage processing: implications for repair and arrest. Science
270:1488–1491. Bell, S. B., R. KobayashiandB. Stillman, 1993 Yeast origin
recog-nition complex functions in transcription silencing and DNA Mayer, M. L., S. P. Gygi, R. AebersoldandP. Hieter, 2001 Iden-tification of RFC(Ctf18p, Ctf8p, Dcc1p): an alternative RFC com-replication. Science262:1844–1849.
Boulet, A., M. Simon, G. Faye, G. A. BauerandP. M. J. Burgers, plex required for sister chromatid cohesion inS. cerevisiae.Mol. Cell7:959–970.
1989 Structure and function of theSaccharomyces cerevisiae CDC2
gene encoding the large subunit of DNA polymerase III.EMBO Pflumm, M. F., andM. R. Botchan, 2001 Orc mutants arrest in metaphase with abnormally condensed chromosomes. Develop-J.8:1849–1855.
Rieder, C. L., R. W. Cole, A. KhodjakovandG. Sluder, 1995 The chromosome triggers a cell-cycle checkpoint. Mol. Cell7:705– 713.
checkpoint delaying anaphase in response to chromosome
mono-orientation is mediated by an inhibitory signal produced by unat- Weinert, T. A., andL. H. Hartwell, 1993 Cell cycle arrest of cdc mutants and specificity of the RAD9 checkpoint. Genetics134:
tached kinetochores. J. Cell Biol.130:941–948.
Rose, M. D., F.Winstonand P. Hieter, 1989 Laboratory Course 63–80.
Manual for Methods in Yeast Genetics. Cold Spring Harbor Labora- Weinert, T. A., G. L. Kiser andL. H. Hartwell, 1994 Mitotic
tory Press, Cold Spring Harbor, NY. checkpoint genes in budding yeast and the dependence of mitosis Skibbens, R. V., L. B. Corson, D. KoshlandandP. Hieter, 1999 on DNA replication and repair. Genes Dev.8:652–665.
Ctf7p is essential for sister chromatid cohesion and links mitotic Wells, A. E., andA. W. Murray, 1996 Aberrantly segregating cen-chromosome structure to the DNA replication machinery. Genes tromeres activate the spindle assembly checkpoint in budding
Dev.13:307–319. yeast. J. Cell Biol.33:75–84.
Stern, B. M., andA. W. Murray, 2001 Lack of tension at kineto- Yan, H., S. I. GibsonandB. Tye, 1991 Mcm2 and Mcm3, two pro-chores activates the spindle checkpoint in budding yeast. Curr. teins important for ARS activity, are related in structure and
Biol.11:1462–1467. function. Genes Dev.5:944–957.
van Brabant, A. J., C. D. Buchanan, E. Charbonneau, W. L.