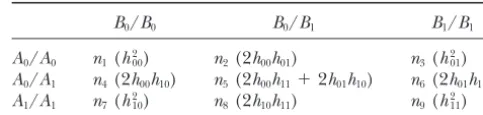DOI: 10.1534/genetics.103.023044
Simultaneous Detection of Linkage Disequilibrium and
Genetic Differentiation of Subdivided Populations
Shuichi Kitada*
,1and Hirohisa Kishino
†*Faculty of Marine Science, Tokyo University of Marine Science and Technology, Minato, Tokyo 108-8477, Japan and †Graduate School of Agricultural and Life Sciences, University of Tokyo, Bunkyo, Tokyo 113-8657, Japan
Manuscript received March 20, 2003 Accepted for publication December 26, 2003
ABSTRACT
We propose a new method for simultaneously detecting linkage disequilibrium and genetic structure in subdivided populations. Taking subpopulation structure into account with a hierarchical model, we estimate the magnitude of genetic differentiation and linkage disequilibrium in a metapopulation on the basis of geographical samples, rather than decompose a population into a finite number of random-mating subpopulations. We assume that Hardy-Weinberg equilibrium is satisfied in each locality, but do not as-sume independence between marker loci. Linkage states remain unknown. Genetic differentiation and linkage disequilibrium are expressed as hyperparameters describing the prior distribution of genotypes or haplotypes. We estimate related parameters by maximizing marginal-likelihood functions and detect linkage equilibrium or disequilibrium by the Akaike information criterion. Our empirical Bayesian model analyzes genotype and haplotype frequencies regardless of haploid or diploid data, so it can be applied to most commonly used genetic markers. The performance of our procedure is examined via numerical simulations in comparison with classical procedures. Finally, we analyze isozyme data of ayu, a severely exploited fish species, and single-nucleotide polymorphisms in humanALDH2.
W
ITH the many discoveries of fine-scale markers developed a method called the transmission/disequilib-rium test (TDT), which uses nuclear family data to de-that represent highly polymorphic loci, linkagedis-tect real associations in structured populations. To use equilibrium between these markers and disease genes
this type of reliable information, similar family-based or other trait genes has regained importance (Jorde
methods for testing linkage disequilibrium have been 1995). Along with the rapid progress of genetic
tech-developed (e.g.,ExcoffierandSlatkin1998;
Lazze-niques, assessing linkage disequilibrium has become a
roni and Lange 1998; Spielman and Ewens 1998).
current concern. The assessment involves two aspects:
However, family-based methods cannot be used for asso-detecting the presence of disequilibrium and estimating
ciation studies of undomesticated species, such as wild-its magnitude once disequilibrium has been confirmed
life and forest trees, for which no nuclear family records (Weir 1979). Continuous efforts have been made on
are available (WuandZeng2001). Hence, population-such assessments of linkage disequilibrium between
al-based methods that detect linkage disequilibrium even leles at two or more loci (Hill1974a,b; Brown1975;
in the presence of population structure are required.
Weir andCockerham 1978;Slatkin and Excoffier
Pritchard et al. (2000a,b) proposed a
population-1996;Luo1998;LuoandSuhai1999;Luoet al.2000;
based method that can detect associations between
AyresandBalding2001;LuoandWu2001). Methods
marker alleles and phenotypes in structured popula-for linkage disequilibrium-based mapping of target
tions. The essential idea of the method is to decompose genes have also been developed (HillandWeir1994;
a sample drawn from a mixed population into several
Kaplanet al.1995;XiongandGuo1997;Meuwissen
unstructured subpopulations and test the association in
andGoddard2000;WuandZeng2001). These
meth-the homogeneous subpopulations. The methods have ods assume homogeneous natural populations.
How-been applied to association analyses in humans (Parra
ever, if a population is structured, this leads to biased
et al.1998;Rosenberget al.2002) and crop plants, with results (known as spurious association) that can reject
modified test statistics being used to deal with quantita-a null quantita-associquantita-ation between quantita-a phenotype quantita-and moleculquantita-ar
tive traits (Thornsberryet al.2001). markers (e.g.,LanderandSchork1994).
To study the population structure, a sample is often To overcome this problem, Spielman et al. (1993)
divided by geographical regions. However, in many cases, there are no obvious regional units by which subpopula-tions can be defined. Rather, natural populasubpopula-tions have
1Corresponding author:Tokyo University of Marine Science and
Tech-a complex hierTech-archicTech-al structure, forming Tech-a metTech-apopu-
metapopu-nology, 4-5-7 Konan, Minato, Tokyo 108-8477, Japan.
E-mail: [email protected] lation (PannellandCharlesworth2000). In models,
a metapopulation has a continuous structure and con- lations that have reached equilibrium under the joint effects of drift and mutation or migration, Wright
sists of an infinite number of subpopulations, or demes.
Such populations are well described by hierarchical (1945, 1951) found that allele frequencies for loci with two alleles have a beta distribution; for multiallele loci, models that specify the distribution of genetic structure
among demes. Population subdivision (NeiandLi1973; the distribution is Dirichlet. We assumed that hyperpar-ameters are common over subpopulations and the sum
Petersonet al.1999) and other evolutionary forces such
as genetic drift (HillandRobertson1968), natural se- of the hyperparameters is also common for all loci. So, if random sampling of demes is performed from a lection (Lewontin1964), and mutation (Ohta1982a,b)
affect linkage disequilibrium. Therefore, when assess- metapopulation, estimated hyperparameters describe ing linkage disequilibrium in natural populations, it is the magnitude of genetic differentiation among sub-very important to also estimate the magnitude of genetic populations that have reached equilibrium. The mean
differentiation. linkage disequilibrium coefficient over subpopulations
In this article, we propose a new strategy to detect is also written as a function of the hyperparameters, as linkage disequilibrium between marker loci and simulta- shown later. Therefore, our assumption on hyperpara-neously estimate genetic differentiation in metapopula- meters has genetic meaning.
tions, extending the empirical Bayesian method of Now,Kdemes are randomly sampled from the
meta-Kitadaet al.(2000). Instead of decomposing a popula- population, and nk individuals are randomly sampled
tion into a finite number of subpopulations, we describe from each deme (k⫽ 1, . . . ,K). Given the allele fre-a distribution for fre-allele frequencies within subpopulfre-ations quencies at each deme, the sample counts of alleles, and test linkage disequilibrium using an information nk⫽ (nk1, . . . ,nkI)⬘, follow a multinomial distribution.
criterion based on hierarchical models that detect either Taking account of uncertainty of the allele frequen-linkage equilibrium or disequilibrium. Using samples cies, the likelihood of the sample counts is expressed from randomly sampled localities from a metapopulation, as a marginal-likelihood function, which is a Dirichlet-we estimate hyperparameters associated with linkage multinomial distribution (e.g., Lange 1995; Rannala
disequilibrium and genetic differentiation, on the basis and Hartigan 1996; Weir 1996; Kitada et al. 2000; of marginal-likelihood functions derived from prior dis- Balding2003):
tributions of allele or haplotype frequencies and
likeli-L˜(␣|nk)⫽
冮
. . .冮
L(p|nk)(p|␣)dphood functions. We assume that individuals mate ran-domly and that Hardy-Weinberg (H-W) equilibrium holds
in each locality or deme. The method can be applied to ⫽ nk!
兿
nki!⌫()
⌫( ⫹n)
兿
I
i⫽1
⌫(␣i⫹nki)
⌫(␣i)
. (2)
frequency data of common genetic markers, including
isozymes, mtDNA, microsatellites, and single-nucleotide The total likelihood is the product of the likelihoods polymorphisms (SNPs). We do not assume independence
ofKdemes. Here,nk⫽
兺
Ii⫽1nki.between marker loci, and linkage states remain unknown.
With a sample size ofn, the variance-covariance ma-We examine the performance of our procedure via
nu-trix of a Dirichlet-multinomial distribution is (n⫹ )/ merical simulations in comparison with classical
proce-(1⫹ ) times larger than that of the multinomial distri-dures. Finally, we analyze isozyme data of ayu, a severely
bution (JohnsonandKotz1969). This phenomenon, exploited fish species, and single-nucleotide
polymor-in which the variance exceeds the nompolymor-inal variance, is phisms in humanALDH2.
called overdispersion. In our hierarchical models, over-dispersion corresponds to the variation of allele fre-quencies over subpopulations. With samples of size
MODELS AND METHODS
n1, . . ,nk, our overdispersion,2, becomes
Distribution of allele frequencies and genetic
differ-entiation:We consider a metapopulation that consists 2 ⫽n⫹
1⫹ . (3)
of localities or demes. Hardy-Weinberg equilibrium holds in each deme. The distribution of the allele frequencies,
Here,nis the mean sample count overKsamples given p⫽(p1, . . . ,pI)⬘, at each deme is described as a
Dirich-byn⫽
兺
Kk⫽1nk/K. For a panmictic population,2 is 1;
let distribution (JohnsonandKotz1969),
it takes values⬎1 according to the magnitude of genetic differentiation among subpopulations. On the other
(p|␣)⫽ ⌫()
兿
Ii⫽1⌫(␣i)
兿
Ii⫽1
p␣ii⫺1, (1)
hand,converges to infinity for a panmictic population. From Weir’s equation (Weir 1996, p. 48, Equation whereIis the number of alleles and ⫽
兺
Ii⫽1␣iand␣⫽ 2.15), the variance of the allele frequency between
sub-(␣1, . . . , ␣I)⬘ are regarded as hyperparameters for re- populations is expressed as
spective alleles specifying prior distribution. We use this
distribution as a prior for allele frequencies of a subdi- Var[p]⫽ p(1⫺p)
popu-TABLE 1
The termFST(2n⫺1)⫹1 corresponds to the dispersion parameter2. From this,Kitadaet al.(2000) obtained
Notation for number of observed composite genotypes
the relation between2andF
ST(Wright1951):
B0/B0 B0/B1 B1/B1
FST⫽ 2⫺1
2n⫺ 1. (4) A0/A0 n1(h2
00) n2(2h00h01) n3(h201) A0/A1 n4(2h00h10) n5(2h00h11⫹2h01h10) n6(2h01h11)
If the organism is haploid, 2nshould ben. By substitut- A1/A1 n7(h2
10) n8(2h10h11) n9(h211) ing Equation 3 into Equation 4 and assumingn⫽2n, we
Diplotype probabilities are given in parentheses.
obtain the relation
FST⫽ 1
1⫹ , (5) lations have a Dirichlet (for casesIj ⬎ 2) or beta (for
casesIj⫽2) distribution. The marginal-likelihood
func-which is also given inBalding(2003). From Equation tion for a subpopulation is obtained from Equations 1,
5, we have 2, and 7 as
⫽ 1
FST
⫺1. (6) L˜0(␣|n)⫽C⬘2nhetero
兿
J
j⫽1
冦
⌫()
⌫( ⫹2n)
兿
Ij
i⫽1
⌫(␣(j)
i ⫹2n(ij))
⌫(␣(ij))
冧
, (8)
This coincides with Equation 3 ofRannalaand
Harti-where 2n(j)
i , the number of genes for allelei at locusj, gan(1996), which was proposed byWright(1969) and
is calculated from composite genotypes. The total likeli-gives the rate of gene flow. Thus, the sum of
hyperpara-hood is the product ofKlikelihood functions for respec-meters is consistent with the rate of gene flow and
samples. This equation can also be used for haploid has a relation withFST.
organisms by usingniandCinstead of 2niandC⬘2nhetero.
Linkage equilibrium with population structure:When
Linkage disequilibrium with population structure:
genetic data are available from multiple loci, the
likeli-The likelihood for sample haplotypes of a subpopula-hood is obtained as a product of the likelisubpopula-hoods of
tion is also a multinomial distribution: these loci when they are under linkage equilibrium. We
consider diploid organisms. Let the frequency allele i L
1(p|n)⫽C
兿
i(1)...i(J)
(hi(1)...i(J))ni (1)...i(J)
. (9)
in a subpopulation at locusjbep(j)
i (j⫽1, . . . ,J;i⫽
1, . . . ,Ij), whereJis the number of loci. The haplotype
If the haplotypes are observed, the marginal likelihood frequency of the subpopulation under linkage
equilib-is given by rium can be written by the product of allele frequencies
over the loci as
L˜1(␣|n)⫽C
⌫()
⌫( ⫹n)
兿
I
i⫽1
⌫(␣i⫹ni)
⌫(␣i)
, (10)
hi(1) ...i(J)⫽p
(1)
i . . .p(iJ),
whereIis the number of haplotypes and, for simplicity, where the combination ofJallelesi(1). . .i(J)should be
we use a suffix i for haplotypes instead of i(1) . . . i(J).
i(1)
j . . .i(jJ), but we use ellipses for simplicity. Let the
sam-However, when analyzing diploid data, we do not ob-ple count for haplotypes be ni(1)
...i(J) and 兺ni(1)...i(J)⫽2n
serve diplotypes (and then no haplotypes), but only (individuals). The likelihood for sample haplotypes
composite genotypes. under H-W equilibrium is a multinomial distribution,
When investigating linkage disequilibrium, the link-age phase between two alleles may be of most interest. L0(p|n)⫽C
兿
i(1)...i(J)
(p(1)
i · · ·p(iJ))ni
(1)
...i(J)⫽C
兿
J
j⫽1
兿
IJ
i⫽1 (p(j)
i )2n
(j) i ,
Hence, here we focus on models for two loci with two alleles and derive the general marginal-likelihood func-where 2n(j)
i is the number of genes of alleleiat locus
tion in theappendix. j. The constant termC ⫽2n!/⌸ini(1)...i(J)! is the
combina-Let allele frequencies for two loci be pA0,pA1andpB0,
tion of the observed haplotypes. However, we do not
ob-pB1, and let the haplotype frequencies beh00,h01,h10,h11in serve haplotypes but genotypes. When composite
geno-a subpopulgeno-ation. In this cgeno-ase, nine composite genotypes typesGi(1)...i(J)are observed, the likelihood is written as
can be observed with specific diplotypes. Let the number L0(p|n)⫽C⬘2nhetero
兿
i(1)...i(J) (pGi(1)
...i(J))
nGi(1)...i(J), (7) of individuals for the genotypes ben
1, . . . ,n9(Table 1). For example, the diplotype for the composite genotype A0A0B1B1can be specified byA0B1/A0B1. Under the H-W whereC⬘ ⫽n!/⌸i(1)...i(J)nGi(1)
...i(J)! andnheterois the sum of the
equilibrium assumption, the probability of having the heterozygous loci overn individuals. Under H-W
equi-genotype ish2
01and that for A0B0/A0B1 is 2h00h01. Thus, librium, probabilities for heterozygous genotypes are
the probabilities of having genotypes can be written by obtained by duplicating the product of allele
frequen-diplotype probabilities, which are combinations of hap-cies. The term 2nheterorefers to such duplication.
subpopu-two diplotypes are possible for the genotypes. In this
DˆTOTAL⫽ ␣
ˆ00␣ˆ11⫺ ␣ˆ01␣ˆ10
ˆ2 . (14)
case,A0A1B0B1is the double heterozygote with observed number n5. For this genotype, the possible diplotypes
However, this estimator is biased. LetE[D] be the mean are A0B0/A1B1 and A0B1/A1B0. The probability for the
ofDat each deme, which can be written as genotype is then given as 2h00h11⫹2h01h10(Table 1).
The likelihood of composite genotypes for the case E[D]⫽E[h00⫺pA
0pB0]
of two loci with two alleles is given in Hudson (2001,
⫽E[h00]⫺E[pA0]E[pB0]⫹E[pA0]E[pB0]⫺E[pA0pB0]
p. 316, Equation 11.6). Expanding the term for the double
heterozygote, we obtain the likelihood function as ⫽
DTOTAL⫺Cov[pA0,pB0],
from which we have L1(p|n)⫽C⬘2n2⫹n4⫹n5⫹n6⫹n8
兺
n5
t⫽0
冢
n5t
冣
(h00)2n1⫹n2⫹n4⫹n5⫺t(h01)n2⫹2n3⫹n6⫹t
DTOTAL⫽E[D]⫹ Cov[pA0,pB0]. (15) ⫻(h10)n4⫹2n7⫹n8⫹t(h11)n5⫹n6⫹n8⫹2n9⫺t, (11)
AspA0⫽h00⫹ h01andpB0 ⫽h00⫹h10, the covariance is whereC⬘ ⫽n!/⌸9
i⫽1ni! is a constant term for the combina- expressed as
tion of the multinomial likelihood. The
marginal-likeli-Cov[pA0,pB0]⫽Var[h00]⫹Cov[h00,h10]⫹Cov[h00,h01] hood function for a subpopulation is then obtained as
⫹ Cov[h01,h10]. L˜1(␣|n)⫽C⬘2n2⫹n4⫹n5⫹n6⫹n8兺
n5
t⫽0
冢
n5
t
冣
⌫()⌫( ⫹2n)
The variance and covariance of a Dirichlet distribution are Var[pi]⫽ ␣i(i⫺ ␣i)/(2( ⫹1)) and Cov[pi,pj]⫽ ⫻ ⌫(␣00⫹2n1⫹n2⫹n4⫹n5⫺t)
⌫(␣00)
⌫(␣01⫹n2⫹2n3⫹n6⫹t)
⌫(␣01) ␣i␣j/(2( ⫹1)) (JohnsonandKotz1969). Hence, we
have the covariance as a function of hyperparameters: ⫻ ⌫(␣10⫹n4⫹2n7⫹n8⫹t)
⌫(␣10)
⌫(␣11⫹n5⫹n6⫹n8⫹2n9⫺t)
⌫(␣11)
,
Cov典 [pA0,pB0]⫽ ␣
ˆ00␣ˆ11⫺ ␣ˆ01␣ˆ10
ˆ2(ˆ ⫹ 1) . (16) (12)
where␣11⫽ ⫺ ␣00⫺ ␣01⫺ ␣10. From Equations 14, 15, and 16, we have the unbiased
Parameter estimation and model selection: We esti- estimator of the mean linkage disequilibrium coefficient
mate parameters by maximizing the negative log mar- correcting spurious association as ginal-likelihood functions for linkage equilibrium and
disequilibrium. The constant terms C and C⬘ can be Eˆ[D]⫽ ␣ˆ00␣ˆ11⫺ ␣ˆ01␣ˆ10
ˆ(ˆ ⫹ 1) , (17)
excluded from the estimation procedure. We
reparame-terize hyperparameters with FST by using the relation while correction ofris not trivial because of the denomi-given by Equation 6 and estimateFSTand hyperparamet- nator of Equation 13.
ers numerically as free parameters by using simplex We use the Akaike Information Criterion (AIC;Akaike minimization. The rate of gene flowand the dispersion 1973) as a criterion for model selections,
parameter2are then estimated by Equations 4 and 6.
AIC⫽2⫻LL典 ⫹2⫻ u, (18) Our empirical Bayesian procedure thus offers
maxi-mum-likelihood estimators of genetic differentiation. where LL典 is the maximum marginal log-likelihood of We estimate the 95% confidence interval for FSTfrom the model anduis the number of free parameters esti-the log-likelihood profile, and esti-then estimate 95% con- mated. We can compare various models by this criterion; fidence intervals forand2by substituting the lower
the model with the lowest AIC value is selected as the and upper confidence limits ofFSTinto Equations 4 and most parsimonious model. Equation 18 indicates that,
6, respectively. when there are several models with similar values of the
We also estimate the linkage correlation coefficient maximum likelihood, we should select the model with
(HillandRobertson 1968) as the smallest number of parameters, again following the
principle of parsimony. By using AIC, we can detect link-age equilibrium or disequilibrium.
rˆ⫽ DˆTOTAL
冪
Eˆ[pA0](1⫺ Eˆ[pA0])Eˆ[pB0](1⫺Eˆ[pB0]), (13)
TESTING PERFORMANCE
whereE[pA0] andE[pB0] are mean allele frequencies over To evaluate the performance of our method, we con-ducted two sets of simulations. The first simulation ex-subpopulations, which are estimated from sample allele
rium with two other commonly used estimates: an esti- erated from the Dirichlet distribution, where the hyper-parameters were given as␣00⫽ (0.64⫹0.16r), ␣01⫽ mate from a pooled sample and an average of the
esti-␣10 ⫽ 0.16(1 ⫺ r), and ␣11 ⫽ (0.04 ⫹ 0.16r). We mated linkage disequilibrium over subsamples.
set total sample size to 1000 individuals and generated
Simulated data 1:We assumed that the mean
popula-haplotype frequencies for different numbers of locali-tion allele frequencies of the two loci over
subpopula-ties (K⫽ 10, 20, 40, 100) under various r(0, 0.2, 0.4, tions wereE[pA0]⫽E[pB0]⫽0.5 and hence haplotype
0.6, 0.8) with fixedFST⫽0.2. We then calculated sample frequencies were E[h00] ⫽ E[h11] ⫽ 0.25(1 ⫹ r) and
composite genotype counts on the basis of haplotype E[h01]⫽E[h10]⫽0.25(1⫺r). We set the sample size to
frequencies assuming H-W equilibrium. 50 individuals for each sampling point and generated
We estimatedFST,E[D], and hyperparameters on the haplotype frequencies for 20 geographical samples
un-basis of Equations 8, 12, and 17 for 1000 replicates. der variousFST(0, 0.05, 0.10, 0.15, 0.20) andr(0, 0.2,
DMEAN and DPOOL were estimated by the method of Hill 0.4, 0.6, 0.8). We then calculated sample composite
(1974b) on the basis of sample composite genotype genotype counts on the basis of haplotype frequencies
counts at each locality and the pooled counts. A larger assuming H-W equilibrium.
number of sampling points improved the precision of For the state of linkage equilibrium (r ⫽ 0), given
estimates ofFST(Table 3). Our hierarchical model esti-theFSTvalues, sample allele frequencies of two loci were
mated the real values ofE[D] correctly over the whole generated independently from the beta distribution
range of linkage disequilibrium and numbers of
sam-(/2,/2), where ⫽1/FST⫺1. Haplotype
frequen-pling points. Precision of the estimates ofE[D] was also cies were then calculated asE[pA0]E[pB0] forE[h00]. Other
improved for a larger number of sampling points (Table haplotypes were calculated in a similar way. For the case
4). Estimates ofDMEAN were biased for weaker linkage ofFST⫽0, sample allele frequencies of the two loci were
disequilibrium. The biases were decreased for largerr; generated independently from the binomial
distribu-however, the biases were larger than those from the tion Bi(50, 0.5). For linkage disequilibrium, given the
hierarchical model. Estimates ofDPOOLwere largely
bi-FSTvalues, haplotype frequencies were generated from
ased with low precision over the whole range of linkage the Dirichlet distributionD(␣00,␣01,␣10,␣11), where the
disequilibrium and numbers of sampling points. The hyperparameters were given as␣00⫽ ␣11⫽ 0.25(1 ⫹
results showed that our method works more efficiently r) and␣01⫽ ␣10⫽ 0.25(1⫺ r). For cases ofFST⫽0,
than classical procedures. sample haplotype frequencies were generated from the
multinomial distribution Mul(50,E[h00], E[h01],E[h10],
E[h11]). APPLICATION TO REAL DATA
We estimatedFST,r, and hyperparameters on the basis Genotype data of the ayu:We analyzed isozyme geno-of Equations 8, 12, and 13 for 1000 replicates. For all
type data for seven samples of the ayu (Plecoglossus alti-cases with r⬎ 0, the model of linkage disequilibrium
velis) from Japan (K. Yoshizawa, unpublished data). had smaller values of AIC and was selected. On the
Two samples of wild stocks were taken from Biwako Lake other hand, for all cases ofr⫽0, the model of linkage
(land-locked type) and Nagara River (amphidoromous equilibrium was selected. Estimates obtained from the
type), and five samples were taken from captive brood best-fit model agreed well with the real values ofFSTand stocks in hatcheries. The lifespan of ayu is 1 year. These
r, showing that our method works appropriately over a brood stocks have been bred in captivity for between 7 wide range of genetic differentiation and linkage dis- and 26 generations in each hatchery to produce juve-equilibrium (Table 2). niles for release to enhance severely exploited stocks.
Simulated data 2: In an ordinary way, researchers The numbers of generations of captive breeding are
would use the sample mean of the linkage disequilib- shown in parentheses with the names of the sampling rium coefficient in each subpopulation (say,DMEAN) as locations in the Table 5 legend. Two loci were analyzed. an estimator of the mean linkage disequilibrium coeffi- Four alleles were found at theGpilocus and three alleles cient. Although a larger number of sampling points at theMpilocus. However, two alleles ofGpiand one of should improve precision of estimates of genetic differ- Mpiwere very minor, and so we grouped them with major entiation, limited sample size in each locality may result ones to obtain nine composite genotypes (Table 5). in a poor estimate ofDMEAN. Another procedure is to esti- We estimated parameters on the basis of composite mate the mean linkage disequilibrium coefficient from genotype data by using Equations 8, 12, and 13 (Table 6). pooled genotype data over subpopulations (say,DPOOL). A smaller AIC value was obtained for the model of link-This procedure neglects the population structure. age equilibrium; thus, linkage equilibrium betweenGpi The mean population allele frequencies of the two loci andMpiloci was detected. Estimates ofFSTandshowed over subpopulations were assumed asE[pA0]⫽E[pB0]⫽0.8 very large genetic differentiation and small gene flow.
Generally, genetic differentiation of fish species is small and hence haplotype frequencies were E[h00]⫽ 0.64 ⫹
0.16r,E[h01]⫽E[h10]⫽0.16(1⫺r), andE[h11]⫽0.04⫹ because of large gene flows caused by migration or lack of barriers (e.g.,McPhersonet al.2001;Goldand
gen-TABLE 2
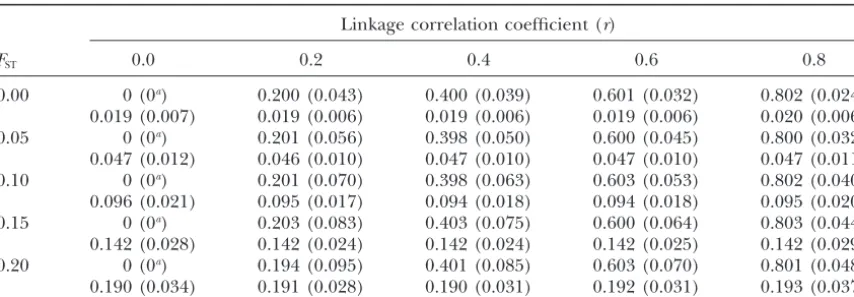
Mean linkage correlation coefficientsrandFSTof the best-fit models estimated from 1000 simulations
under various levels of linkage disequilibrium and population structure
Linkage correlation coefficient (r)
FST 0.0 0.2 0.4 0.6 0.8
0.00 0 (0a) 0.200 (0.043) 0.400 (0.039) 0.601 (0.032) 0.802 (0.024)
0.019 (0.007) 0.019 (0.006) 0.019 (0.006) 0.019 (0.006) 0.020 (0.006)
0.05 0 (0a) 0.201 (0.056) 0.398 (0.050) 0.600 (0.045) 0.800 (0.032)
0.047 (0.012) 0.046 (0.010) 0.047 (0.010) 0.047 (0.010) 0.047 (0.011)
0.10 0 (0a) 0.201 (0.070) 0.398 (0.063) 0.603 (0.053) 0.802 (0.040)
0.096 (0.021) 0.095 (0.017) 0.094 (0.018) 0.094 (0.018) 0.095 (0.020)
0.15 0 (0a) 0.203 (0.083) 0.403 (0.075) 0.600 (0.064) 0.803 (0.044)
0.142 (0.028) 0.142 (0.024) 0.142 (0.024) 0.142 (0.025) 0.142 (0.029)
0.20 0 (0a) 0.194 (0.095) 0.401 (0.085) 0.603 (0.070) 0.801 (0.048)
0.190 (0.034) 0.191 (0.028) 0.190 (0.031) 0.192 (0.031) 0.193 (0.037)
The sample size was set to 50 individuals for each sampling point and haplotype frequencies for 20 geographi-cal samples were generated. The mean allele frequencies of the two loci were assumed asE[pA0]⫽E[pB0]⫽0.5. The numbers in parentheses are the standard errors of the estimates.
aIn all replicates, the model of linkage equilibrium was selected.
ner2002). However, the result obtained here is natural to 0.01 for 17 subpopulations, with an average of⫺0.45
(Petersonet al.1999, Table 4). Strong linkage
disequi-because the sample comprised three different groups:
land-locked, amphidoromous, and captive brood stocks librium was found, which is consistent with the analyses by Petersonet al. Estimates ofFSTandalso showed that in successive breeding for many generations.
Human SNPs:We analyzed SNPs reported byPeter- genetic differentiation of humanALDH2was large with
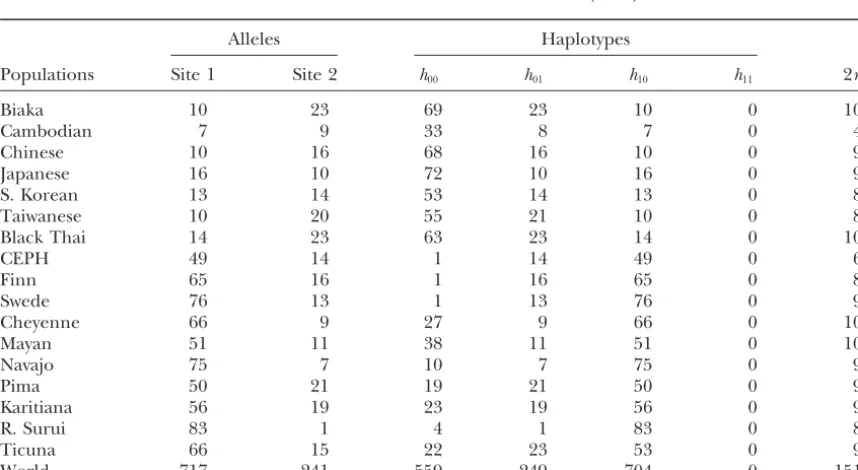
a small gene flow.
sonet al.(1999) in the human aldehyde dehydrogenase 2 geneALDH2.
We focused on sites 1 and 2 from the six sites reported
DISCUSSION
by Petersonet al.The haplotype frequencies were
esti-mated from the haplotype configurations given in their We have proposed a new method, based on genotype frequencies of geographical samples, to detect linkage Table 2 ash11⫽ H1 ⫹ H4 ⫹ H6 ⫹ H8 ⫹ H9, h12⫽ H3,
h21⫽H2, and h22⫽ 0. We estimated parameters using disequilibrium between markers and to simultaneously estimate genetic differentiation by taking population Equation 8 from the allele frequencies at the two sites
withinALDH2genotyped in 756 people from 17 popula- subdivision into account. The method detected linkage disequilibrium and estimated linkage disequilibrium co-tions across five continents (Table 7). We also estimated
parameters for estimated haplotypes on the basis of efficient andFSTcorrectly.
With a hierarchical model, we estimate the genetic Equation 10 (Tables 7 and 8). The AIC value for the
model of linkage disequilibrium was smaller than that structure in a metapopulation, rather than decompose a population into a finite number of randomly mating for linkage equilibrium. Our estimate of r was ⫺0.60,
whereas the original authors’ estimates varied from⫺0.96 subpopulations. The sum of hyperparameterscoincided
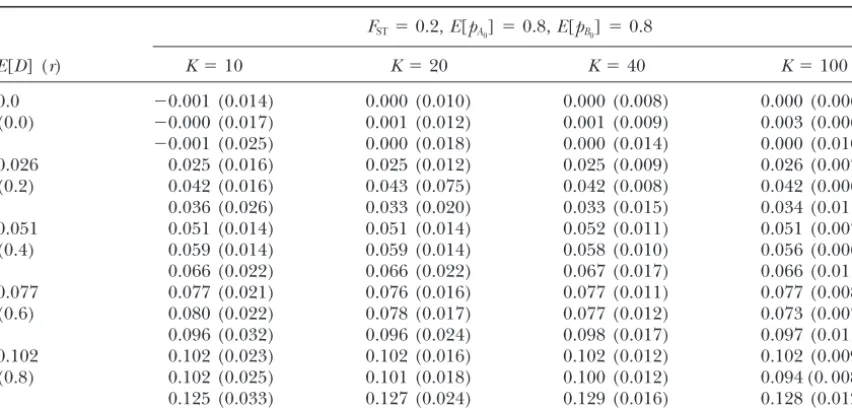
TABLE 3
MeanFSTestimated from 1000 simulations under various levels of linkage disequilibrium
for different numbers of sampling points
FST⫽0.2,E[pA0]⫽0.8,E[pB0]⫽0.8
r K⫽10 K⫽20 K⫽40 K⫽100
0 0.180 (0.047) 0.193 (0.036) 0.195 (0.027) 0.198 (0.020)
0.2 0.181 (0.048) 0.193 (0.037) 0.196 (0.028) 0.198 (0.021)
0.4 0.180 (0.049) 0.190 (0.036) 0.195 (0.028) 0.198 (0.022)
0.6 0.183 (0.052) 0.192 (0.039) 0.197 (0.030) 0.198 (0.023)
0.8 0.183 (0.056) 0.191 (0.044) 0.197 (0.034) 0.198 (0.025)
TABLE 4
Performance of the hierarchical model in comparison with classical procedures
FST⫽0.2,E[pA0]⫽0.8,E[pB0]⫽0.8
E[D] (r) K⫽10 K⫽20 K⫽40 K⫽100
0.0 ⫺0.001 (0.014) 0.000 (0.010) 0.000 (0.008) 0.000 (0.006)
(0.0) ⫺0.000 (0.017) 0.001 (0.012) 0.001 (0.009) 0.003 (0.006)
⫺0.001 (0.025) 0.000 (0.018) 0.000 (0.014) 0.000 (0.010)
0.026 0.025 (0.016) 0.025 (0.012) 0.025 (0.009) 0.026 (0.007)
(0.2) 0.042 (0.016) 0.043 (0.075) 0.042 (0.008) 0.042 (0.006)
0.036 (0.026) 0.033 (0.020) 0.033 (0.015) 0.034 (0.011)
0.051 0.051 (0.014) 0.051 (0.014) 0.052 (0.011) 0.051 (0.007)
(0.4) 0.059 (0.014) 0.059 (0.014) 0.058 (0.010) 0.056 (0.006)
0.066 (0.022) 0.066 (0.022) 0.067 (0.017) 0.066 (0.011)
0.077 0.077 (0.021) 0.076 (0.016) 0.077 (0.011) 0.077 (0.008)
(0.6) 0.080 (0.022) 0.078 (0.017) 0.077 (0.012) 0.073 (0.007)
0.096 (0.032) 0.096 (0.024) 0.098 (0.017) 0.097 (0.011)
0.102 0.102 (0.023) 0.102 (0.016) 0.102 (0.012) 0.102 (0.009)
(0.8) 0.102 (0.025) 0.101 (0.018) 0.100 (0.012) 0.094 (0. 008)
0.125 (0.033) 0.127 (0.024) 0.129 (0.016) 0.128 (0.012)
Total sample size was fixed at 1000 individuals. The numbers are means estimated from 1000 simulations with standard errors in parentheses. Lines are arranged in threes as follows: top (E[D]), mean of the linkage correlation coefficient at each deme estimated by the hierarchical model; middle (DMEAN), sample mean of the linkage disequilibrium coefficient in each deme; bottom (DPOOL), estimates of the linkage disequilibrium coefficient based on pooled genotypes over demes.
with the rate of gene flow, andFSTwas written by. We territory. Even for marine fish, which can disperse widely because of the lack of barriers in oceans, the stock con-also showed by Equation 17 that the magnitude of
link-age disequilibrium depends on that of genetic differen- cept is popular in fishery resource management (Waples
1998). In breeding seasons, fish species generally gather tiation. Our model assumes metapopulations, which
in-cludes Wright’s island model as a special case (Wright to spawning grounds where they mate randomly. As a result of such breeding patterns, the assumption of 1940); hence the relationship betweenandFSTcan be
applied for these population models. The metapopula- Hardy-Weinberg equilibrium may be valid in each local-ity. Furthermore, in humans, whose population struc-tion concept fits with ecology of wildlife that have
lim-ited movement ability and may mate randomly in their tures may be more complex than those of wildlife, it has
TABLE 5
Allozyme composite genotype frequencies for seven samples of the ayu from Japan
Sampling location
Genotypes GM-1a GM-2a NGa GFb SGc HSa OTa
A0A0B0B0 1 1 65 32 4 3 79
A0A0B0B1 0 2 25 8 7 2 0
A0A0B1B1 0 0 1 2 3 0 0
A0A1B0B0 9 9 7 41 14 30 30
A0A1B0B1 17 18 2 5 27 23 0
A0A1B1B1 9 7 0 2 16 5 0
A1A1B0B0 18 17 0 7 5 19 2
A1A1B0B1 35 36 0 3 18 16 0
A1A1B1B1 11 10 0 1 6 2 0
Total 100 100 100 100 100 100 111
GM, Gunma (26); NG, Niigata (7); GF, Gifu; SG, Shiga; HS, Hiroshima (21); and OT, Oita (11) Prefecture. Numbers in parentheses are the numbers of generations in captive breeding.
aCaptive brood stocks.
bWild (amphidoromous type).
TABLE 8 TABLE 6
Estimated linkage correlation coefficient between Estimated worldwide linkage correlation coefficient between
sites 1 and 2 in the humanALDH2gene and genetic
GpiandMpiloci and genetic differentiation
among seven samples of ayu differentiation among 17 human populations
Models Models
Linkage Linkage
Linkage Linkage
equilibrium disequilibrium equilibrium disequilibrium
LLa ⫺1444.79 ⫺1247.85
LLa ⫺1073.25 ⫺1093.89
AIC 2152.50 2195.78 AIC 2895.58 2503.70
r — ⫺0.601
r — 0.024
FST 0.280 [0.168, 0.455] 0.209 FST 0.204 0.234 [0.166, 0.295]
3.908 3.267 [2.390, 5.024]
b 2.576 [1.198, 4.952] 3.787
2c 29.164 [17.920, 46.825] 22.039 2 18.917 21.608 [15.598, 26.943]
aMaximum log-likelihood without the multinomial con-aMaximum log likelihood without the multinomial constant
termC⬘in Equations 8 and 12. stant termCin Equations 8 and 10.
bRate of gene flow.
cDispersion parameter.
this treatment was appropriate. Consider a simple case of three haplotypes with sample countsn1,n2, and n3, been reported that geographic clusters often correspond where n
3 ⫽ 0. The marginal-likelihood function for a closely to predefined regional or population groups or subpopulation is a Dirichlet-multinomial distribution: collections of geographically similar populations (
Rosen-L1(␣1,␣2,␣3|n1,n2,n3)⫽ ⌫
(␣1⫹ ␣2⫹ ␣3)
⌫(␣1⫹ ␣2⫹ ␣3⫹n1⫹n2)
⌫(␣1⫹n1)
⌫(␣1)
⌫(␣2⫹n2)
⌫(␣2)
.
berget al.2002). In actual populations, various levels of
subdivision may exist (Excoffier2001), and the num- (19)
ber of demes should be very large with continuous
sub-Using the relation of⌫(n⫹1)⫽n⌫(n), the first term division. In ecological studies, sampling points may
in-of this equation can be written as crease year by year, which increases the accuracy of our
method. ⌫(␣1⫹ ␣2⫹ ␣3)
⌫(␣1⫹ ␣2⫹ ␣3)(␣1⫹ ␣2⫹ ␣3⫹1) . . . (␣1⫹ ␣2⫹ ␣3⫹n1⫹n2⫺1)
Estimates of hyperparameters were based only on
fre-quencies counted in the samples, and alleles that did not ⫽{(␣1⫹ ␣2⫹ ␣3⫹1) . . . (␣1⫹ ␣2⫹ ␣3⫹n1⫹n2⫺1)}⫺1.
(20) appear were assigned frequency 0. Here, we consider if
TABLE 7
Haplotype frequencies at two sites (sites 1 and 2) in the humanALDH2gene among 17 human populations,
calculated from Table 2 ofPetersonet al.(1999)
Alleles Haplotypes
Populations Site 1 Site 2 h00 h01 h10 h11 2n
Biaka 10 23 69 23 10 0 102
Cambodian 7 9 33 8 7 0 48
Chinese 10 16 68 16 10 0 94
Japanese 16 10 72 10 16 0 98
S. Korean 13 14 53 14 13 0 80
Taiwanese 10 20 55 21 10 0 86
Black Thai 14 23 63 23 14 0 100
CEPH 49 14 1 14 49 0 64
Finn 65 16 1 16 65 0 82
Swede 76 13 1 13 76 0 90
Cheyenne 66 9 27 9 66 0 102
Mayan 51 11 38 11 51 0 100
Navajo 75 7 10 7 75 0 92
Pima 50 21 19 21 50 0 90
Karitiana 56 19 23 19 56 0 98
R. Surui 83 1 4 1 83 0 88
Ticuna 66 15 22 23 53 0 98
Hudson, R. R., 2001 Linkage disequilibrium and recombination,
Hence, to maximize Equation 19, the term in the
paren-pp. 309–324 in Handbook of Statistical Genetics, edited by D. J.
theses in Equation 20 should be minimized. As␣3ⱖ0, Balding, M.Bishopand C.Cannings. Wiley, Chichester, UK.
Johnson, N. L., and S.Kotz, 1969 Discrete Distributions. Wiley, New
the maximum-likelihood estimate of␣3 is then 0. This
York.
can be generalized for all cases.
Jorde, L. B., 1995 Linkage disequilibrium as a gene mapping tool.
For diploid data, we have two procedures for inferring Am. J. Hum. Genet.56:11–14.
Kaplan, N., W. G. HillandB. S. Weir, 1995 Likelihood methods
linkage disequilibrium. The first procedure (method 1)
for locating disease genes in nonequilibrium populations. Am.
estimates hyperparameters using Equation 12 or the last
J. Hum. Genet.56:18–32.
equation in the appendix, on the basis of observed Kitada, S., T. HayashiandH. Kishino, 2000 Empirical Bayes
proce-dure for estimating genetic distance between populations and
composite genotypes. Another way (method 2) is to
effective population size. Genetics156:2063–2079.
use Equation 10, on the basis of estimated haplotypes.
Lander, E. S., andN. J. Schork, 1994 Genetic dissection of complex
Several methods for estimating haplotype frequencies traits. Science265:2037–2048.
Lange, K., 1995 Application of the Dirichlet distribution to forensic
have been developed (Excoffier and Slatkin 1995;
match probabilities. Genetica96:107–117.
Longet al.1995;SlatkinandExcoffier1996). In the
Lazzeroni, L. C., and K. Lange, 1998 A conditional inference
analysis of human SNPs, we used estimated haplotypes framework for extending the transmission/disequilibrium test.
Hum. Hered.48:67–81.
as if they were observed data. However, the effect of
us-Lewontin, R. C., 1964 The interaction of selection and linkage. I.
ing estimated haplotypes on parameter estimates is
un-General considerations; heterotic models. Genetics49:4–67.
known. The accuracy of the inference might depend on Long, J. C., R. C.Williamsand M.Urbanek, 1995 An E-M algorithm
and testing strategy for multiple-locus haplotypes. Am. J. Hum.
the precision of haplotype estimates. Method 1 should
Genet.56:799–810.
be better for accurate inference, but deriving explicit
Luo, Z. W., 1998 Detecting linkage disequilibrium between a
poly-marginal-likelihood functions becomes much harder morphic marker locus and a trait locus in natural populations.
Heredity80:198–208.
for larger numbers of loci, although the general form
Luo, Z. W., andS. Suhai, 1999 Estimating linkage disequilibrium
of the marginal-likelihood function is derived in the
between a polymorphic marker locus and a trait locus in natural
appendix. Method 2 is much easier and more practical. populations. Genetics151:359–371.
Luo, Z. W., andC.-I Wu, 2001 Modeling linkage disequilibrium
Precise estimation of haplotype frequencies is very
im-between a polymorphic marker locus and a locus affecting
com-portant for studies of linkage disequilibrium.
plex dichotomous traits in natural populations. Genetics 158:
1785–1800. We thank Kazutomo Yoshizawa for allowing us to use his
unpub-Luo, Z. W., S. H. TaoandZ-B. Zeng, 2000 Inferring linkage
disequi-lished data and Laurent Excoffier and an anonymous reviewer for their
librium between a polymorphic marker locus and a trait locus constructive comments on earlier versions of the manuscript. This
in natural populations. Genetics156:457–467.
work was supported by the Japan Society for the Promotion of Science
McPherson, A. A., R. L. Stephenson, P. T. O’Reilly, M. W. Jones
and Japan Science and Technology Agency.
andC. T. Taggart, 2001 Genetic diversity of coastal Northwest
Atlantic herring population implications for management. J. Fish
Biol.59:356–370.
Meuwissen, T. H. E., andM. E. Goddard, 2000 Fine mapping of
LITERATURE CITED
quantitative trait loci using linkage disequilibria with closely
linked marker loci. Genetics155:421–430.
Akaike, H., 1973 Information theory and an extension of the
maxi-mum likelihood principle, pp. 267–281 inThe Second International Nei, M., and W.-H. Li, 1973 Linkage disequilibrium in subdivided
populations. Genetics75:213–219.
Symposium on Information Theory, edited by B. N.Petrovand F.
Csa´ki. Akade´miai Kiado´, Budapest. Ohta, T., 1982a Linkage disequilibrium due to random genetic
drift in finite subdivided populations. Proc. Natl. Acad. Sci. USA
Ayres, K. L., andD. J. Balding, 2001 Measuring gametic
disequilib-rium from multilocus data. Genetics157:413–423. 79:1940–1944.
Ohta, T., 1982b Linkage disequilibrium with the island model.
Ge-Balding, D. J., 2003 Likelihood-based inference for genetic
correla-tion coefficient. Theor. Popul. Biol.63:221–230. netics75:139–155.
Pannell, J. R., andB. Charlesworth, 2000 Effects of
metapopula-Brown, A. D. H., 1975 Sample sizes required to detect linkage
dis-equilibrium between two or three loci. Theor. Popul. Biol.8:184–201. tion processes on measures of genetic diversity. Philos. Trans. R.
Soc. Lond. B355:1851–1864.
Excoffier, L., 2001 Analysis of population subdivision, pp. 271–307
inHandbook of Statistical Genetics, edited by D. J. Balding, M. Parra, E. J., A. Marcini, J. Akey, J. Maartinson, M. A. Batzeret al.,
1998 Estimating African American admixture proportions by
Bishopand C.Cannings. Wiley, Chichester, UK.
Excoffier, L., andM. Slatkin, 1995 Maximum likelihood estima- use of population-specific alleles. Am. J. Hum. Genet.63:1839–
1851. tion of molecular haplotype frequencies in a diploid population.
Mol. Biol. Evol.12:921–927. Peterson, R. J., D. GoldmanandJ. C. Long, 1999 Effects of
world-wide population subdivision onALDH2linkage disequilibrium.
Excoffier, L., andM. Slatkin, 1998 Incorporating genotypes of
relatives into a test of linkage disequilibrium. Am. J. Hum. Genet. Genet. Res.9:844–852.
Pritchard, J. K., M. Stephens, N. A. RosenbergandP. Donnelly,
62:171–180.
Gold, J. R., andT. F. Turner, 2002 Population structure of red drum 2000a Association mapping in structured populations. Am. J.
Hum. Genet.67:170–181.
(Sciaenops ocellatus) in the northern Gulf of Mexico, as inferred
from variation in nuclear-encoded microsatellites. Marine Biol.140: Pritchard, J. K., M. StephensandP. Donnelly, 2000b Inference
of population structure using multilocus genotype data. Genetics 249–265.
Hill, W. G., 1974a Tests for association of gene frequencies at sev- 156:945–959.
Rannala, B., andJ. A. Hartigan, 1996 Estimating gene flow in
eral loci in random diploid populations. Biometrics31:881–888.
Hill, W. G., 1974b Estimation of linkage disequilibrium in randomly island populations. Genet. Res.67:147–158.
Rosenberg, N. A., J. K. Pritchard, J. L. Weber, H. M. Cann, K. K.
mating populations. Heredity33:229–239.
Hill, W. G., andA. Robertson, 1968 Linkage disequilibrium in Kiddet al., 2002 Genetic structure of human populations.
Sci-ence298:2381–2384.
finite populations. Theor. Appl. Genet.38:226–231.
Hill, W. G., andB. S. Weir, 1994 Maximum likelihood estimation Slatkin, M., andL. Excoffier, 1996 Testing for linkage
disequilib-rium in genotype data using the EM algorithm. Heredity76:377–383.
of gene location by linkage disequilibrium. Am. J. Hum. Genet.
in the presence of association: the sib
transmission/disequilib-rium test. Am. J. Hum. Genet.62:450–458. L1⫽ C⬘
兿
m
i⫽1
冢
兺
lGi
j⫽1
PDj
冣
nGi.
Spielman, R. S., R E. McGinnisandW. J. Ewens, 1993 Transmission
test for linkage disequilibrium: the insulin gene region and
insu-lin-dependent diabetes mellitus (IDDM). Am. J. Hum. Genet.52: By multinomial expansion, we have another form of the
506–513. likelihood as
Thornsberry, J. M., M. M. Goldman, J. Doebley, S. Kresovich,
D. Nielsenet al., 2001 Dwarf8 polymorphisms associate with
variation in flowering time. Nat. Genet.28:286–289.
L1⫽ C⬘
兺
i1...ilG1
nG1!
i(1)1 ! . . .i(1)lG 1!
冢
Pi
(1) 1
D(G1)
1 . . .P
il(1)
G1 D(G1)
lG1
冣
. . .
Waples, R. S., 1998 Separating the wheat from the chaff. Pattern
of genetic differentiation in high gene flow species. J. Hered.89:
438–450.
Weir, B. S., 1979 Inferences about linkage disequilibrium.
Biomet-兺
i1...il
Gm
nGm!
i(1m)! . . .i (m)
lGm!
冢
Pi(m) 1
D(Gm) 1 . . .P
i(lGmm)
D(Gm) lGm
冣
,
rics35:235–254.
Weir, B. S., 1996 Genetic Data Analysis II. Sinauer Associates,
Sunder-land, MA.
Weir, B. S., andC. C. Cockerham, 1978 Testing hypotheses about
wherei(i)
j is the number of individuals having diplotype
linkage disequilibrium with multiple alleles. Genetics88:633–642.
j, which constitutes genotypei, andd兺lGi
j⫽1i(
i)
j ⫽nGi.
Wright, S., 1940 Breeding structure of populations in relation to
speciation. Am. Nat.74:232–248. Under H-W equilibrium, the probability of the jth
Wright, S., 1945 The differential equation of the distribution of
diplotype is expressed as a product of the probabilities
gene frequencies. Proc. Natl. Acad. Sci. USA31:383–389.
of haplotypes,
Wright, S., 1951 The general structure of populations. Ann. Eugen.
15:323–354.
Wright, S., 1969 Evolution and Genetics of Populations: The Theory of Ph
j1/hj2⫽ 2
(1⫺␦hj1hj2)P hj1Phj2,
Gene Frequencies, Vol. 2. University of Chicago Press, Chicago.
Wu, R., andZ-B. Zeng, 2001 Joint linkage and linkage disequilibrium
where ␦ is a delta function that is 1 for homozygous
mapping in natural populations. Genetics157:899–909.
Xiong, M. M., andS. W. Guo, 1997 Fine-linkage genetic mapping and 0 for heterozygous genotypes. Therefore, the
likeli-based on linkage disequilibrium theory and applications. Am. J. hood of a sample from a subpopulation under H-W
Hum. Genet.60:1513–1531.
equilibrium is
Communicating editor: L.Excoffier
L1⫽C⬘
兺
i1...ilG1
nG1!
i(1)1 ! . . .i (1) lG
1
!冦
共
2(1⫺␦h(G1) 11h(G1) 12
) P(G1)
h11 P
(G1) h12
兲
i(1) 1
. . .
APPENDIX: GENERAL LINKAGE DISEQUILIBRIUM
MARGINAL-LIKELIHOOD FUNCTION
共
2(1⫺␦h(G1)lG1
h(G1)
lG12)P(G1)
hl
G1
1P (G1) hl
G12
兲
i(1)
lG1
冧
. . .兺
i1...il
Gm
nGm!
i(1m)! . . .i (m) lG
m!
FOR DIPLOID ORGANISMS
Let observed composite genotypes be Gi(i ⫽ 1, . . . , ⫻
冦
共
2(1⫺␦h(Gm) 11h
(Gm) 12
) P(Gm)
h11 P (Gm)
h12
兲
i(m)1
. . .
共
2(1⫺␦h(Gm)lGm1hl(GmGm2))P(Gm)
hl
Gm
1P (Gm)
hl
Gm
2
兲
i(m)lGm
冧
. m) and the numbers of individuals for the genotypes benGi. The likelihood function for a sample is written as In the above equation some haplotypes will be common,
so we rename these haplotypes as L1⫽C⬘
兿
m
i⫽1
PnGi
Gi,
{h⬘1, . . . ,h⬘}⫽{h(
G1) 11 , . . . ,h
(Gm)
lGm2},
where PGiis the probability for the genotypeGi, which
whereis the number of common haplotypes. The like-can be written by the probability of diplotypes. For the
lihood function is then written as case of two loci with two alleles, the diplotype
probabili-ties are given in Table 1. Even for multilocus data, a
diplotype can be specified for a set of completely homo- L1⫽C⬘
兺
i1...ilG1
. . .
兺
i1...il
Gm
nG1!
i(1)1 ! . . .i (1)
lG1!
. . . nGm!
i(1m)! . . .i (m)
lGm!
zygous genotypes, but the larger number of
heterozy-gous genotypes results in a larger number of diplotype ⫻
冦
2(1⫺␦h(G1) 11 h(G1) 12
)
冧
i(1) 1. . .
冦
2(1⫺␦h(lGm)Gm1h (Gm)
l
Gm2
)
冧
i(m)lGm
(Ph⬘i)
j1. . . (P
h⬘)j,
combinations. Consequently, the number of summations of diplotype probabilities increases for such genotypes.
where js (s ⫽ 1, . . . , ) is the number of individuals
Thus, the probability of a genotype is expressed by the
having haplotypesand兺js⫽2n,
sum of diplotype probabilities.
Let a set of diplotypes and haplotypes consistent with j
s⫽i
(1) 1
共
␦h⬘sh(G1) 11 ⫹ ␦h⬘sh
(G1)
12
兲
⫹. . .⫹i(1)
lG1
共
␦h⬘sh(G1)
lG11 ⫹ ␦h⬘sh
(G1)
lG12
兲
⫹. . .a genotypeGbe
⫹i(1m)
共
␦h⬘sh(Gm) 11 ⫹ ␦h⬘sh
(Gm)
12
兲
⫹. . .⫹i(m)
lGm
共
␦h⬘sh(Gm)
lGm1 ⫹ ␦h⬘sh
(Gm)
lGm2
兲
,diplo(G)⫽ (D(1G), . . . ,D(
G)
lG )⬘
where␦h⬘sh(11G1)is again a delta function that is 1 ifh⬘s
coin-⫽ (h(11G)/h(
G)
12, . . . ,h(
G)
lG1/h(
G)
lG2)⬘,
cides withh(G1)
11 and 0 otherwise. Thus, the likelihood for where lG is the number of diplotypes consistent with observed composite genotypes can be written as the sum
of the multinomial distribution for haplotype frequen-the genotypeG. The likelihood function for composite
cies. From aboveL1, the marginal-likelihood function genotypes is then written by using diplotype
L˜1(␣|n)⫽ C⬘
兺
i1...ilG 1
. . .
兺
i1...il
Gm
nG1!
i(1)1 ! . . .i(1)lG 1!
. . . nGm! i(1m)! . . .i(
m)
lGm! ⫻
冦
⌫()
⌫( ⫹2n)
兿
s⫽1
⌫(␣s⫹js)
⌫(␣s)
冧
.
The total marginal-likelihood function is obtained
mul-⫻
冦
2(1⫺␦h(G1)11 h(12G1))
冧
i(1)1
. . .
冦
2(1⫺␦h(Gm) lGm1h(lGmGm2))冧
i(lGmm)