Targeting gastrointestinal stromal tumors: the role of regorafenib
Full text
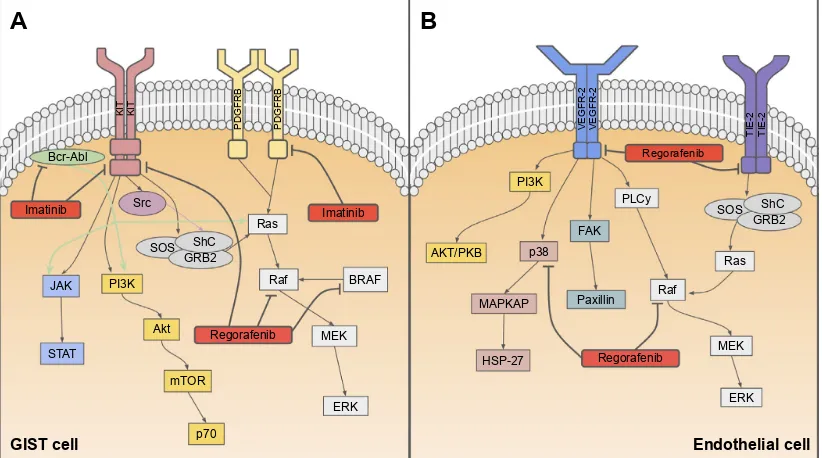
Figure

Related documents
The patients were submitted to a treatment regimen for 1.5 hours per day adapted for cellulite, consisting of manual and mechanical lymph drainage and cervical stimulation using
that, by inhibiting the activity of the BET protein BRD4 and the resulting inhibition of c-Myc, co-treatment with AS and JQ1 attenuated the expression of several prosurvival genes,
According to the findings of this study, the factors that correlated with the fertility desire among PLHIV were age, current health status, having a sexual partner during the
Such classrooms will also be engaging places since understanding core ideas requires experiences with the practices (Michaels et al., 2008). Interestingly, one item from the
Aim: The present study was aimed at investigating the role of literacy and generation in the self- reported general health status of Moroccan Berber speaking women in the
The adjuvant suspension consists of small submicron particles that form continuous porous surfaces, and dense surface texture, which may impact antigen
The subscales mental health, role difficulty, and ocu- lar pain had a similar pattern of correlations between the patients’ responses to the questionnaire items by subscales and
In rheumatoid arthritis, there occurs a failure of acute inflammation to resolve may predispose to: autoimmunity, chronic dysplastic inflammation, excessive
