Acta Cryst.(2001). E57, o1089±o1090 DOI: 10.1107/S1600536801017548 Bond and Davies C6H7N
o1089
organic papers
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
2-Picoline
Andrew D. Bond* and John E. Davies
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, England
Correspondence e-mail: [email protected]
Key indicators Single-crystal X-ray study T= 120 K
Mean(C±C) = 0.003 AÊ Disorder in main residue Rfactor = 0.062 wRfactor = 0.181
Data-to-parameter ratio = 10.5
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
The crystal structure of 2-picoline (2-methylpyridine, C6H7N)
has been determined at 120 (2) K following in situ crystal growth from the liquid. Molecules pack in a herring-bone-type arrangement in the non-centrosymmetric space groupP212121,
with CÐH N contacts indicative of directional hydrogen bonds.
Comment
The picolines (methylpyridines) comprise a series of empirical formula C6H7N, with weak intermolecular interactions and
low melting points. The crystal structure of 4-picoline (4-methylpyridine; m.p. 276 K) has been determined previously from a crystal grown using an elaborate modi®ed Bridgman technique (Ohms et al., 1985). We report here the crystal structure of 2-picoline (m.p. 206 K), determined at 120 (2) K from a crystal grown in situ in a 0.3 mm glass capillary. This work forms part of a study devoted to improving techniques for determining the crystal structures of substances that are liquids at room temperature (see, for example, Bond & Davies, 2001).
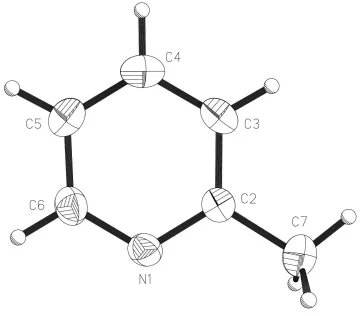
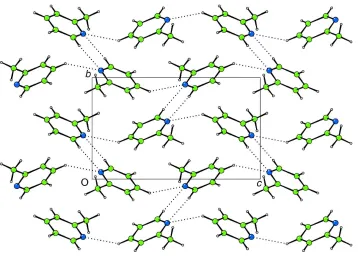
Molecules of (I) (Fig. 1) pack in a herring-bone-type arrangement (Fig. 2) in the non-centrosymmetric space group
P212121. Between molecules, CÐH N contacts exist that are
close to linear [H4 N1i= 2.60 AÊ and C4ÐH4 N1i= 146.6;
H6 N1ii = 2.79 AÊ and C6ÐH6 N1ii = 170.1; symmetry
codes: (i)1
2ÿx, ÿy, 12+z; (ii)ÿ12+x, ÿy, ÿz], indicative of
directional hydrogen bonds.
Experimental
The sample (99%) was obtained from the Aldrich Company and was used without further puri®cation. The crystal was grown in a 0.3 mm glass capillary tube at 206 K (a temperature only slightly less than the melting point of the solid in the capillary tube) using a technique described previously (Davies & Bond, 2001). The crystal was subse-quently cooled to 120 (2) K for data collection. The length of the cylindrical crystal was not estimated, but it exceeded the diameter of the collimator (0.35 mm).
Crystal data C6H7N Mr= 93.13
Orthorhombic,P212121 a= 6.6593 (5) AÊ
b= 7.0878 (6) AÊ
c= 11.7358 (7) AÊ
V= 553.93 (7) AÊ3 Z= 4
Dx= 1.117 Mg mÿ3
MoKradiation Cell parameters from 4571
re¯ections = 1.0±27.5 = 0.07 mmÿ1 T= 120 (2) K Cylinder, colourless 0.15 mm (radius)
Data collection
Nonius KappaCCD diffractometer Thin-slice!and'scans 3477 measured re¯ections 749 independent re¯ections 672 re¯ections withI> 2(I)
Rint= 0.110 max= 27.5 h=ÿ4!8
k=ÿ9!7
l=ÿ15!10 Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.062 wR(F2) = 0.181 S= 1.10 749 re¯ections 71 parameters
H atoms treated by a mixture of independent and constrained re®nement
w= 1/[2(F
o2) + (0.1425P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.29 e AÊÿ3
min=ÿ0.29 e AÊÿ3
The methyl H atoms are disordered and were modelled as two sets of idealized positions. All H atoms were placed geometrically and allowed to re®ne with independent isotropic displacement
para-meters (one common parameter for all methyl H atoms). The methyl group was allowed to rotate about its local threefold axis. Friedel pairs (486) were averaged before merging of data in P212121; the
reported value of Rint corresponds to subsequent merging of
equivalent re¯ections in this space group.
Data collection:COLLECT(Nonius, 1998); cell re®nement:HKL SCALEPACK(Otwinowski & Minor, 1997); data reduction: HKL DENZO (Otwinowski & Minor, 1997) and SCALEPACK; program(s) used to solve structure: SIR92 (Altomare et al., 1994); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); software used to prepare material for publication:SHELXL97.
We thank the EPSRC for ®nancial assistance towards the purchase of the Nonius CCD diffractometer.
References
Altomare, A., Cascarano, G., Giacovazzo, C., Guagliardi, A., Burla, M. C., Polidori, G. & Camalli, M. (1994).J. Appl. Cryst.27, 435±436.
Bond, A. D. & Davies, J. E. (2001).Acta Cryst.E57, o1039±o1040. Davies, J. E. & Bond, A. D. (2001).Acta Cryst.E57, o947±o949. Nonius (1998).COLLECT. Nonius BV, Delft, The Netherlands.
Ohms, U., Guth, H., Treutmann, W., DannoÈhl, H., Schweig, A. & Heger, G. (1985).J. Chem. Phys.83, 273±279.
Otwinowski, Z. & Minor, W. (1997). Methods in Enzymology, Vol. 276,
Macromolecular Crystallography, Part A, edited by C. W. Carter & R. M. Sweet, pp. 307±326. London: Academic Press.
Sheldrick, G. M. (1993).XP. University of GoÈttingen, Germany. Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany. Watkin, D. J., Prout, C. K. & Pearce, L. J. (1996).CAMERON. Chemical
Crystallography Laboratory, University of Oxford, England. Figure 2
Projection onto (100) showing the herring-bone packing in (I) (CAMERON; Watkinet al., 1996). CÐH N interactions are shown as dotted lines.
Figure 1
supporting information
sup-1
Acta Cryst. (2001). E57, o1089–o1090
supporting information
Acta Cryst. (2001). E57, o1089–o1090 [doi:10.1107/S1600536801017548]
2-Picoline
Andrew D. Bond and John E. Davies
S1. Comment
The picolines (methylpyridines) comprise a series of empirical formula C6H7N, with weak intermolecular interactions and
low melting points. The crystal structure of 4-picoline (4-methylpyridine; m.p. 276 K) has been determined previously
from a crystal grown using an elaborate modified Bridgman technique (Ohms et al., 1985). We report here the crystal
structure of 2-picoline (m.p. 206 K), determined at 120 (2) K from a crystal grown in situ in a 0.3 mm glass capillary.
This work forms part of a study devoted to improving techniques for determining the crystal structures of substances that
are liquids at room temperature (see, for example, Bond & Davies, 2001).
Molecules of (I) (Fig. 1) pack in a herring-bone-type arrangement (Fig. 2) in the non-centrosymmetric space group
P212121. Between molecules, C—H···N contacts exist that are close to linear [H4···N1i = 2.60 Å and C4—H4···N1i =
146.6°; H6···N1ii = 2.79 Å and C6—H6···N1ii = 170.1°; symmetry codes: (i) 0.5 - x, -y, 0.5 + z; (ii) -0.5 + x, -y, -z],
indicative of directional hydrogen bonds.
S2. Experimental
The sample (99%) was obtained from the Aldrich Company and was used without further purification. The crystal was
grown in a 0.3 mm glass capillary tube at 206 K (a temperature only slightly less than the melting point of the solid in the
capillary tube) using a technique described previously (Davies & Bond, 2001). The crystal was subsequently cooled to
120 (2) K for data collection. The length of the cylindrical crystal was not estimated, but it exceeded the diameter of the
collimator (0.35 mm).
S3. Refinement
The methyl H atoms are disordered and modelled as two sets of idealized positions. All H atoms were placed
geometrically and allowed to refine with independent isotropic displacement parameters (one common parameter for all
methyl H atoms). The methyl groups were allowed to rotate about its local threefold axis. Friedel pairs (486) were
merged prior to merging in P212121; the reported value of Rint corresponds to subsequent merging of equivalent reflections
Figure 1
The molecular structure and atom-labelling scheme for (I) showing displacement ellipsoids at the 50% probability level
supporting information
sup-3
[image:5.610.129.487.68.325.2]Acta Cryst. (2001). E57, o1089–o1090 Figure 2
Projection onto (001) showing the herring-bone packing in (I) (CAMERON; Watkin et al., 1996). C—H···N interactions
are shown as dotted lines.
2-methylpyridine
Crystal data C6H7N
Mr = 93.13
Orthorhombic, P212121
a = 6.6593 (5) Å b = 7.0878 (6) Å c = 11.7358 (7) Å V = 553.93 (7) Å3
Z = 4 F(000) = 200
Dx = 1.117 Mg m−3
Melting point: 206.3 K Mo Kα radiation, λ = 0.7107 Å Cell parameters from 4571 reflections θ = 1.0–27.5°
µ = 0.07 mm−1
T = 120 K
Cylinder, colourless 0.15 mm (radius)
Data collection Nonius KappaCCD
diffractometer
Radiation source: fine-focus sealed tube Thin–slice ω and φ scans
3477 measured reflections 749 independent reflections
672 reflections with I > 2σ(I) Rint = 0.110
θmax = 27.5°, θmin = 3.5°
h = −4→8 k = −9→7 l = −15→10
Refinement Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.062
wR(F2) = 0.181
S = 1.10 749 reflections
71 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.29 e Å−3
Δρmin = −0.29 e Å−3
Special details
Experimental. Grown in situ in a 0.3 mm Lindemann capillary tube at 206 K. Freidel pairs (486) were averaged for the refinement.
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq Occ. (<1)
N1 0.3417 (3) 0.0686 (3) 0.05357 (16) 0.0301 (5) C2 0.4756 (3) −0.0479 (3) 0.10232 (18) 0.0275 (6) C3 0.4444 (4) −0.1249 (3) 0.2101 (2) 0.0335 (6) H3 0.5432 −0.2040 0.2437 0.071 (12)* C4 0.2683 (4) −0.0852 (3) 0.26800 (19) 0.0382 (6) H4 0.2440 −0.1368 0.3414 0.067 (10)* C5 0.1293 (4) 0.0306 (3) 0.2168 (2) 0.0356 (6) H5 0.0061 0.0592 0.2539 0.048 (8)* C6 0.1716 (4) 0.1045 (3) 0.1109 (2) 0.0337 (6) H6 0.0751 0.1852 0.0765 0.038 (7)*
C7 0.6606 (4) −0.0923 (4) 0.0342 (2) 0.0408 (7) 0.80 H7A 0.7359 0.0242 0.0201 0.060 (7)* 0.80 H7B 0.6217 −0.1490 −0.0386 0.060 (7)* 0.80 H7C 0.7449 −0.1810 0.0768 0.060 (7)* 0.80 C7′ 0.6606 (4) −0.0923 (4) 0.0342 (2) 0.0408 (7) 0.20 H7A′ 0.6776 0.0026 −0.0257 0.060 (7)* 0.20 H7B′ 0.6468 −0.2174 −0.0004 0.060 (7)* 0.20 H7C′ 0.7781 −0.0910 0.0845 0.060 (7)* 0.20
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-5
Acta Cryst. (2001). E57, o1089–o1090
C7 0.0321 (11) 0.0448 (13) 0.0456 (14) 0.0058 (11) 0.0036 (11) −0.0012 (11) C7′ 0.0321 (11) 0.0448 (13) 0.0456 (14) 0.0058 (11) 0.0036 (11) −0.0012 (11)
Geometric parameters (Å, º)
N1—C6 1.341 (3) C4—H4 0.9500 N1—C2 1.343 (3) C5—C6 1.379 (3) C2—C3 1.394 (3) C5—H5 0.9500 C2—C7 1.502 (3) C6—H6 0.9500 C3—C4 1.384 (3) C7—H7A 0.9800 C3—H3 0.9500 C7—H7B 0.9800 C4—C5 1.375 (4) C7—H7C 0.9800
C6—N1—C2 117.6 (2) C4—C5—C6 118.9 (2) N1—C2—C3 121.9 (2) C4—C5—H5 120.6 N1—C2—C7 116.5 (2) C6—C5—H5 120.6 C3—C2—C7 121.6 (2) N1—C6—C5 123.6 (2) C4—C3—C2 119.5 (2) N1—C6—H6 118.2 C4—C3—H3 120.3 C5—C6—H6 118.2 C2—C3—H3 120.3 C2—C7—H7A 109.5 C5—C4—C3 118.5 (2) C2—C7—H7B 109.5 C5—C4—H4 120.7 C2—C7—H7C 109.5 C3—C4—H4 120.7