Full-Potential KKR Calculations for Lattice Distortion Effect of Point Defect
in bcc-Fe Dilute Alloys, Based on the Generalized-Gradient Approximation
M. Asato
1,+, C. Liu
2, K. Kawakami
3, N. Fujima
2and T. Hoshino
21National Institute of Technology, Niihama College, Niihama 792-8580, Japan 2Graduate School of Engineering, Shizuoka University, Hamamatsu 432-8561, Japan
3Nippon Steel & Sumitomo Metal Corporation, Futtsu 293-8511, Japan
We present theab-initio calculations for the lattice distortion effect of substitutional single impurities, I (=Sc³Ge, Sn), and substitutional two impurities, II and ISn, in bcc-Fe. Sn is a perturbed-angular-correlation (PAC) probe. The calculations are based on the generalized-gradient-approximation in the density-functional formalism and the full-potential Korringa-Kohn-Rostoker (FPKKR) Green’s function method. For single impurities, we show that the available experimental results, such as lattice distortion around the impurities and the atomic volume changes per impurity, are reproduced very well by the present calculations. For two impurities, we clarify the lattice distortion effect on 1st-nearest neighbor (NN) interaction energies of II and ISn pairs in Fe, being a difference between the distortion energies with two impurities located on the infinitely separated sites and on the neighboring sites. We initially show that the lattice distortion effect is very low for the interaction energies between two impurities with a small size-misfit, compared with the host atom, although it becomes high for the interaction energies of two impurities with a large size-misfit. The lattice distortion effect on the II (I=Cr³Zn) interaction energies is less than 0.02 eV, while the lattice distortion effect on the ISn (I=Sc, Ge) interaction energies is greater than 0.2 eV. Secondly, we show that we can improve the agreement with the experimental results for the interaction energies of ISn pairs in bcc-Fe, by taking into account the lattice distortion. Finally, we show that the magnetic interaction is important for the lattice distortion of the ISn (I=Cr, Mn) pairs. The high lattice distortion for ISn pairs (I=Cr, Mn) is partly caused by the antiferromagnetic interaction of impurities I with the 1st-NN host atom on the opposite site of the Sn impurity, resulting in the high energy gain (0.13 eV, 0.06 eV) of ISn interaction. [doi:10.2320/matertrans.M2013391]
(Received October 17, 2013; Accepted May 26, 2014; Published July 4, 2014)
Keywords: iron-based dilute alloys, point defects, ab-initio calculations, KorringaKohnRostoker Green’s function method, density functional theory, generalized-gradient approximation
1. Introduction
Knowledge of the interaction energies of point defects in solids is indispensable for understanding many of the basic physical processes, such as diffusion, short-range order, and segregation.1,2)Most interesting, from a technological point of
view as well as fundamental point of view, are the interatomic interaction energies in alloys, which are essential for under-standing the phase diagrams.3,4) It is now well known that
ab-initio calculations based on the density-functional theory provide accurate data of the bulk properties of materials as well as defect properties.57)Thus,ab-initio calculations are expected to play an important role in material design.
We have shown that the full-potential KorringaKohn Rostoker (FPKKR) Green’s function method for bulk properties and point defects, developed by the Jülich group, combined with the generalized-gradient approximation, accurately reproduces the point-defect energies as well as the bulk properties of metals.813) For example, the
equi-librium lattice parameters, bulk moduli, and mono vacancy formation energies of elemental face-centered-cubic (fcc) and body-centered-cubic (bcc) metals, Li³Au, are reproduced within the errors of 1, 10, and 10% of the experimental values, respectively.8) We showed that the experimental values14) for the interaction energies of impurities with the perturbed-angular-correlation (PAC) probes (Rh, Pd, In, Sn, Sb) in metals are fairly well reproduced even by the present calculations neglecting the lattice distortion caused by the introduction of impurities.15,16) It is noted that the PAC
spectroscopy has the ability to deal with very low probe-atom
concentrations (³10¹8) without restriction for the
temper-ature. The interaction energies of the probe-impurity pairs are determined from the temperature-dependent measurement of the probe-impurity pair concentrations. We also showed that the experimentally known fundamental features in the phase diagrams of the 4d binary alloys H1¹cIc(H=Nb, Mo, Pd, Ag
(major element); I=Zr³Ag (minor element)), such as segregation, solid solution and order,17)may be understood using the sign and magnitude of the 1st-nearest neighbor (NN) impurity-impurity (II) interaction energies in the H metals.9,10) It is noted that the II interaction energies correspond to the 2-body interaction energies in theab-initio real-space cluster expansion approach for the total energies of the ordered and disordered alloys.18,19) We have recently
shown that ourab-initio cluster expansion approach may be quantitatively useful for the study of the atomic structures and magnetism of alloys because the total energies of alloys may be very accurately reproduced by including only low-order terms, such as the 2-body and 3-body interaction energies, although the 2-body interaction is very long-range. For example, the band energy of Ni2MnAl is reproduced
within the error of 1 mRy per atom by our real-space cluster expansion, if the 2-body interaction up to 10th-NN’s is included.20)
In the preceding paper,21) we have shown that the fundamental features of the Fe-based binary alloys FeI (I=Sc³Ge) are understood in the same way. Using the calculations neglecting the lattice distortion caused by the introduction of impurities, we also well reproduced the experimental results for the interaction energies of impurities with the Sn PAC-probe in Fe. It is obvious, however, that the lattice distortion effect becomes important for the interaction
+Corresponding author, E-mail: asato@sci.niihama-nct.ac.jp
energies between two impurities with a large size-misfit, compared with the host atom. It is noted that the distortion effect on the interaction energies is a distortion-energy difference between two (initial and final) states of two impurities, as discussed in Sect. 2.
We can presently carry out the calculations including the lattice distortion effect around the point defects in crystals, however, the calculations for the lattice distortion effect are very time-consuming and cumbersome. Papanikolaou et al.
already calculated the lattice distortion energies of substitu-tional single impurities (Ti³Ge and Zr³Sn) in Cu and showed how the lattice distortion energies becomes high for single impurities with a large size-misfit compared to the host atom.22)We also calculated the lattice distortion energies of a
vacancy in Al and Cu.23)Very recently, using the formalism
given by the Kanzaki model,1,2) we have very accurately
reproduced the experimental results for the relative atomic volume changes per impurity (Sc³Ge) in Fe.24,25) The
available experimental results for the interatomic distances between impurities and the 1st-NN’s in Fe, obtained by the X-ray absorption fine-structure,26) are also reproduced very well. It is noted that the formalism given by the Kanzaki model is very suitable for the present work, because all the parameters in the Kanzaki model, such as the Hellmann Feynman forces and lattice distortion, caused by impurities in Fe, are estimated accurately by the present ab-initio calculations.24)
The purpose of the present paper is to systematically and quantitatively study the lattice distortion effect of substitu-tional single and two impurities in bcc-Fe. In Sect. 2, we describe the calculation method. In Sect. 3, we show the calculated results for the lattice distortion of single impurities I (=Sc³Ge) in bcc-Fe. We also discuss the lattice distortion effect of magnetism of single impurities in bcc-Fe. Although the preliminary results for single impurities in bcc-Fe have been discussed in Ref. 24), the present paper gives a complete discussion for single impurities. In Sect. 4, we show the calculated results for the lattice distortion effect on the interaction energies and magnetism of II pairs in Fe. In Sect. 5, we show the calculated results for the lattice distortion effect on the interaction energies and magnetism of ISn (Sn=PAC-probe) pairs in bcc-Fe. In Sect. 6 we summarize the main results of the present paper.
We concentrated our study on the lattice distortion effect on the interaction energies of 1st-NN II and ISn pairs in Fe because the lattice distortion effect seems to be mostly important for the 1st-NN interaction energies between two large impurities, as shown in the present paper.
2. Calculation Method
If a defect is inserted in a metal, the HellmannFeynman (HF) forces are induced on the host atoms around the defects. The equilibrium positions of the neighboring host atoms are determined by the zero-force condition. In the present study, in order to calculate the HF forces as well as the total energies, we use the full-potential KorringaKohnRostoker Green’s function method and the generalized-gradient approximation in the density functional formalism.8,9,11)
The advantage of the Green function method is that due to
the introduction of the host Green’s function, the embedding of point defects in an otherwise ideal crystal is correctly described, different from the supercell and cluster calcu-lations. It is noted that the deformation of the wave functions due to the defects is delocalized over the whole space, although the potential perturbation due to the defects is localized in the vicinity of the defects because of the screening effect of the free-electron-like sp-electrons in a metal. The practical advantage in using the Green’s function method is to exploit this short-range nature of the defect potential. For example, we found that, in order to obtain the accurate and converged total energy of the defect system without the lattice distortion effect, it is sufficient to self-consistently redetermine only the potentials of the impurities and their neighboring potentials (up to the1st (2nd)-nearest neighbors (NN’s) of the impurities in the case of fcc (bcc) structure), if the total-energy change due to the perturbed wave functions over the infinite space is correctly evaluated using Lloyd’s formula. The short-range nature of the defect potential has been discussed in Ref. 8).
Thenth-NN interaction energy between the two impurities is defined as the total-energy difference between two states: (1) the final state where the two impurity atoms are located at nth-NN sites and (2) the initial state where the impurity atoms are infinitely-separated. Thus, in order to obtain the interaction energies, we must calculate the total energies of both the states. The calculation procedure for the 1st- and 2nd-NN interaction energies between two impurities in Fe, without the lattice distortion effect, are described in Ref. 21). For example, the potentials in the 22-atom cluster were redetermined self-consistently for the 1st-NN interaction energies.
On the other hand, the treatment of the lattice distortion is slightly cumbersome. The lattice distortion energy is defined as the total-energy change caused by the lattice distortion. We must self-consistently redetermine the potentials in the larger region because the potentials of the host atoms surrounding the displaced host atoms may be strongly perturbed. Since in the present studies we take into account the lattice distortion effect on the interaction energies of the 1st-NN II and ISn impurity pairs (I=Sc³Ge) in bcc-Fe, we self-consistently redetermine the potentials in the 84-atom cluster, as shown in Fig. 1(a), which includes all the host atoms up to the 2nd-NN’s of the displaced host atoms around the two impurities. It is noted that the distortion effect on the interaction energy is defined as the distortion-energy difference between two (initial andfinal) states of two impurities, as discussed above. In order to determine the lattice distortion up to the 2nd-NN’s around single impurities, we redetermine the potentials in the 65-atom cluster, being a part of the 84-atom cluster and shown in Fig. 1(b), which includes all the host atoms up to the 2nd-NN’s of the 2nd-NN’s around the impurities. The 65-atom cluster includes all the atoms up to the 6th-shell of the impurity at the center.
3. Calculated Results for Lattice Distortion of Single Impurities (Sc³Ge) in Fe
We systematically discuss the lattice distortion caused by single impurities (Sc³Ge) in bcc-Fe. In Sect. 3.1, we show the calculated results for the HF forces on the host atoms surrounding single impurities and discuss the distance dependence of the HF forces. In Sect. 3.2, we determine the lattice distortion up to the 2nd-shell of single impurities and show that the lattice distortion of the 1st-NN host atoms is dominant. In Sect. 3.3, using the formalism of the Kanzaki model,1,22,24)we reproduce the available experimental results for the relative atomic volume changes per impurity. All the parameters in the Kanzaki model are determined by the present ab-initio calculations. In Sect. 3.4, we discuss the lattice distortion effect of the magnetism of single impurities in Fe.
[image:3.595.311.547.68.388.2]3.1 Distance dependence of HF forces on the host atoms around impurities in bcc-Fe
Figure 2 shows the calculated results for the HF forces on the host atoms at the ideal atomic positions in the vicinity of an impurity up to the 9th-shell (one impurity+136 host atoms) around single impurities. The positive forces mean the outward relaxation around an impurity, while the negative denotes the inward one. It is noted that the HF forces are dominant on the 1st-NN’s and become weaker on the more distant neighbors, although the oscillating behavior due to the screening effect by the 4sp electrons in Fe (transition-metal) is very complicated, differently from the oscillating behavior of impurities in Al (simple metal).27,28) Thus, in the present
study, we treat only the lattice distortion up to the 2nd-shell around the single impurities. We alsofind that the HF forces on the 5th-NN ð1;1;1Þ, being located on the line of the impurityð0;0;0Þto the 1st NNð0:5;0:5;0:5Þ, become slight stronger for I=Cr³Zn. These may be important for the large expansion caused by a Zn impurity in Fe compared to those caused by Ga and Ge impurities in Fe, as discussed in Sect. 3.3.
3.2 Lattice distortion around single impurities in bcc-Fe We first consider only the lattice distortion of the 1st-NN host atoms,fixing all other atoms at their ideal host positions. Figure 3 shows the lattice distortion energies, the HF forces exerted on the 1st-NN’s, the magnetic moments of the
(a)
(b)
Fig. 1 (a) 84-atom cluster of C3v-symmetry, including two impurities with
the surrounding 82 Fe atoms and (b) 65-atom cluster of Oh-symmetry,
including one impurity with the surrounding 64 Fe atoms, being a part of the 84-atom cluster.
[image:3.595.74.260.70.512.2]impurities, and those of the 1st-NN host atoms as a function of the change of the 1st-NN distance. The lattice distortion energy is defined as the total-energy change caused by the lattice distortion. The positions of the force-zero agree very
well with those of the minima in the lattice distortion energies, as seen in Fig. 3. This result demonstrates the accuracy of the presentab-initio calculations. Our results for the atomic displacements around Cr and Mn are in very good
Fig. 3 Lattice distortion energies (lower panel, parabolic behavior), radial forces (lower panel, line) on a 1st-NN host atom, magnetic moments (upper panel, two lines) of impurities I (I=Sc³Ge), and those of the NN host atom, as a function of the change of the 1st-NN distance. The available experimental results26)for the local lattice distortion around Cr (¹0.30.4%) and Mn (¹0.60.3%) impurities
[image:4.595.144.451.65.669.2]agreement with the available experimental results from extended X-ray absorption fine-structure,26) as shown by
the transverse bars in Fig. 3 (Cr and Mn). The calculated results for the lattice distortion are summarized in Table 1(a). The calculated results for the magnetic moments in Fig. 3 will be discussed in Sect. 3.4.
We next consider both the lattice distortion of the 1st- and 2nd-NN’s. Since the HF forces on the 2nd-NN’s are weak, we can easily obtain the force-zero on the 1st- and 2nd-NN’s by starting from the atomic positions of force-zero on the 1st-NN’s. The calculated results are listed in Table 1(b). It is obvious that the displacement of the 1st-NN host atoms is dominant and does not change very much by taking into account the relaxation of the 2nd-NN’s.
3.3 Relative atomic volume changes per impurity in Fe-based dilute alloys
A lattice expansion or compression due to a single impurity results in a change of the lattice constant. In the case of cubic metals, using the harmonic approximation, the atomic volume change due to an impurity is given by thefirst moment of the Kanzaki forces (FKn)1,22)
V ¼VV0¼31K
X
n Fn
KRn ð1Þ
where V and V0 are the atomic volumes of the impurity
system and ideal host, respectively, and K is the bulk modulus of pure bcc-Fe. We use the results of V0 and K8)
obtained by the present ab-initio calculation method. The Kanzaki forces
Fn
K¼Fn X
n0 nn0
Sn0 ð2Þ
are the forces that would induce the same local distortions, Sn0, in the host crystal as the direct forces, Fn, cause in the defect systems. nn0 is a difference between the force-constant matricesð;0Þof the systems with and without a defect given by the difference between the slopes of the force curves. In Fig. 4, we present the calculated relative atomic volume changes, ¦V/V0, for all the systems considered in
this study. The calculated results agree very well with the available experimental results. It is obvious that the large volume changes for Sc, Ti, Cu, Zn, Ga, and Ge are due to the significant displacement of the 1st-NN host atoms, as shown in Table 1. We alsofind that the atomic volume changes are
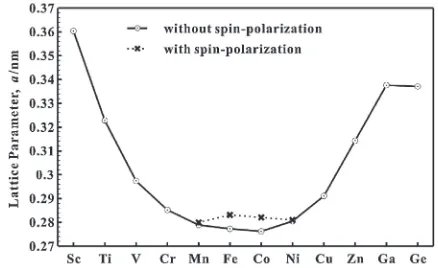
mostly correlated with the equilibrium lattice parameters of metals in the bcc structure, as shown in Fig. 5, which were obtained by the presentab-initio calculations. However, it is noted that the experimentally known significant expansion caused by a Zn impurity, compared to those caused by Ga and Ge impurities, cannot be understood by the above-mentioned simple explanation based on the atomic size effect. It may be understood by comparing the HF forces on the 5th-NN’s in eq. (2), caused by the impurity potential. The HF forces on the 5th-NN’s are stronger for a Zn impurity than for the Ga and Ge impurities, as seen in Fig. 2, and the distance from the center, being an important factor in eq. (1), is two times stronger for the 5th-NN’s than for the 1st-NN’s. Thus, the formalism given by the Kanzaki model is very useful to understand the experimental results for the atomic volume changes caused by single impurities.
3.4 Lattice distortion effect of magnetism of single impurities in bcc-Fe
The changes in the magnetic moments of the impurities and their NN’s, due to the lattice distortion of the 1st-NN’s, are shown in Fig. 3 (upper panel). The magnetic moment (2.16 µB) of 1st-NN Fe does not change very much
[image:5.595.317.535.71.200.2]by the lattice distortion. For magnetic moments of impurities, they are very different, depending on the impurity. We find that the antiferromagnetic coupling to the bulk magnetization of Fe atoms is stable for the early 3d impurities (Sc³Mn), while the ferromagnetic coupling is stable for the late 3d impurities. It is well known that the magnetism of the 3d
Table 1 Calculated lattice distortion (in % of interatomic distance), confined only to the 1st-NN host atoms (a), and to the 1st- and 2nd-NN host atoms (b).
(a)
Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge
1st 3.0 1.3 0.2 ¹0.2 0.1 0 0.0 0.3 0.6 1.2 1.3 1.1 (b)
Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge
1st 2.9 1.3 0.2 ¹0.2 0.1 0 0.0 0.3 0.5 1.1 1.2 1.0 2nd 0.4 0.3 0.2 0.0 ¹0.1 0 0.0 ¹0.1¹0.2¹0.3 ¹0.3 ¹0.2
Fig. 4 Relative volume changes per impurity (open circles), together with the available experimental results (black squares).25)
[image:5.595.316.535.244.378.2]metallic alloys is understood by considering the 3d electron numbers per atom; the anitiferromagnetism is very stable for the half-filled d bands.2932) It is also noted that the
magnetism of the impurities becomes strong with the lattice expansion, as seen in Fig. 3 (I=Cr, Mn, Fe), because the interaction of the surrounding host atoms with the impurity becomes weak. For I=Cr and Mn, the negative magnetic moments increase with the lattice expansion, while for I=Fe, the positive magnetic moment increases.
4. Calculated Results for Lattice Distortion Effect on 1st-NN Interaction Energies and Magnetism of Impurity-Impurity (II, I=Sc³Ge) Pairs
We systematically discuss the distortion effect on 1st-NN II (I=Sc³Ge) interaction energies. It has already been shown in Ref. 21) that the fundamental features of the phase diagram of FeI alloys, such as segregation, solid solution, and order, being experimentally known,17)are understood by
[image:6.595.45.552.94.155.2]using the sign and magnitude of II interaction energies without the lattice distortion effect. In the present calcu-lations, the lattice distortion up to the 2nd-NN host atoms is taken into account, as discussed in Sect. 2. It is noted that the lattice distortion effect on the interaction energy is a distortion-energy difference between two (initial and final) states of two impurities, as discussed in Sect. 2.
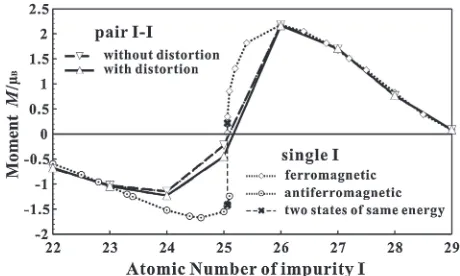
Figure 6 and Table 2 show the calculated results with and without the lattice distortion effect. The changes in the II interatomic distances are also listed in Table 2. It is obvious that the lattice distortion effect on the II interaction energies is very low (<0.02 eV) for I=Cr³Zn with a small size-misfit, but it becomes high for I=Sc³V, Ga, and Ge with a large size-misfit, although it does not change the sign of interaction energies (distinction between attraction and repulsion). We can expect that the chemical trend for atomic sizes of the impurities is similar to that of equilibrium lattice parameters of metals in the bcc structure, as shown in Fig. 5. Thus, the fundamental feature of the present results may be easily understood because the equilibrium lattice parameters of Sc³V, Ga, and Ge are very large compared to that of Fe. The long II (I=Sc³V, Ga, Ge) interatomic distances, being listed in Table 2, may be mainly due to the large atomic size of the impurities. The significant repulsion of II (I=Sc³V) may be understood by considering the breakup of two strong dd (FeI) bonds. It is noted that the II interaction energy is defined as the total-energy difference due to the creation of two bonds (II and FeFe) and the breaking of two bonds (FeI).21) The strength of the d
d-bond for I=Sc³Cu may be understood by the bandfilling of the 3d electrons.2932) On the other hand, the significant
repulsion of II (I=Ga and Ge) may be partly due to the breaking of the strong spd (FeI) bonds.
We now discuss the magnetism of the impurity pairs in bcc-Fe. The magnetic moments of II (I=Sc³Ge) pairs as well as those of the single impurities are shown in Fig. 7. As discussed in Sect. 3.2, the antiferromagnetic (↓) coupling to the bulk magnetization of Fe (↑) is stable for the early 3d-transition metals (Sc³Mn), while the ferromagnetic (↑) coupling is stable for the late 3d-transition metals (Fe³Cu). For two identical impurities in Fe, the ferromagnetic coupling (↓↓) between two impurities, antiparallel to the bulk magnetization of Fe (↑), is stable for I=Sc³Cr, while the ferromagnetic coupling (↑↑) between two impurities, parallel to the bulk magnetization of Fe (↑), is stable for I=Fe³Cu. The change in the magnetic moments, due to the pairing, is
Fig. 6 Calculated interaction energies of 1st-NN II (I=Sc³Ge) pairs in Fe, with and without the lattice distortion effect.
Table 2 Calculated interaction energies (in eV) for the 1st-NN II (I=Sc³Ge) pairs in Fe with and without the lattice distortion effect. The changes in the II interatomic distances are also shown in parentheses (in%).
Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge
With distortion
0.25 (8.3)
0.25 (6.9)
0.22 (4.8)
0.21 (2.6)
¹0.03 (¹2.4)
0 (0)
0.05 (0.1)
¹0.02 (1.2)
¹0.21 (¹0.2)
¹0.12 (1.4)
0.10 (4.9)
0.33 (9.1) Without
distortion 0.35 0.36 0.28 0.23 ¹0.01 0 0.05 ¹0.01 ¹0.19 ¹0.11 0.14 0.48
[image:6.595.315.538.185.320.2] [image:6.595.311.541.367.505.2]very small except for I=Cr and Mn. It is noted that the change in the magnetic moments of Mn is very large. For the MnMn pair, we can obtain two solutions for the magnetism of the 1st- and 2nd-NN interactions (antiferromagnetic (↑↓) and ferromagnetic (↓↓) couplings between two Mn impurities in the bulk magnetization of Fe (↑)), as shown in Fig. 8. Figure 8 shows the distance dependence of MnMn interaction energies. The distance behavior is very compli-cated compared to that of the MnMn interaction energies in Al.27) According to our calculations, the antiferromagnetic coupling (↑↓) between two Mn impurities is stable by 0.076 eV (0.026 eV) for the 1st (2nd)-NN interaction compared to the ferromagnetic coupling (↓↓) between two Mn impurities. For the CrCr pair, on the other hand, we obtain only one solution (↓↓) for the magnetism. These results mean that the antiferromagnetic coupling (↑↓) between two Mn impurities is much stronger than that of FeMn pair. It is also noted that the lattice distortion effect on II (I=Cr, Mn) interaction energies is as low as 0.021 and 0.022 eV for the 1st-NN CrCr (↓↓) and MnMn (↑↓) pairs, respectively, as shown in Fig. 6 and listed in Table 2.
5. Calculated Results for Lattice Distortion Effect on 1st-NN Interaction Energies and Magnetism of Impurity-Sn (ISn, I=Sc³Ge) Pairs in Fe
We systematically discuss the calculated results for the lattice distortion effect on 1st-NN ISn (I=Sc³Ge) interaction energies in Fe. It has already been shown in Ref. 21) that the chemical trend of the experimental results17)
[image:7.595.62.284.103.358.2]is reproduced very well by the calculations neglecting the lattice distortion effect. In the present calculations, the lattice distortion up to the 2nd-NN host atoms around two impurities is taken in to account, as discussed in Sect. 2. It is noted that the lattice distortion effect on the interaction energy is a distortion-energy difference between two (initial and final) states of two impurities, as discussed in Sect. 2.
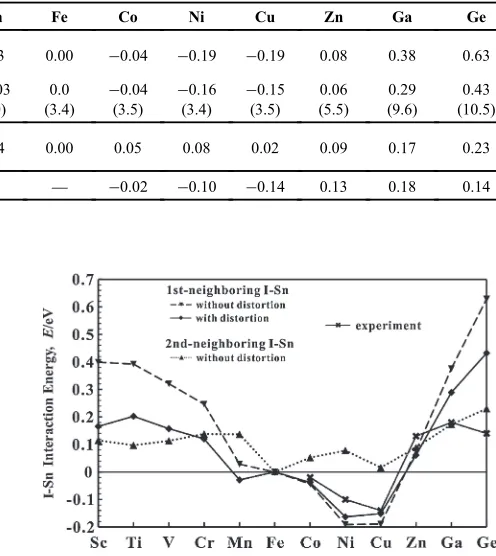
Figure 9 and Table 3 show the calculated results for the ISn interaction energies, with and without the lattice distortion effect. Wefind that we can improve the agreement with the experimental results, by taking into account the lattice distortion. The changes in interatomic distances of ISn pairs are also listed in Table 3. It is obvious that the lattice distortion effect is very high for the interaction energies of ISn (I=Sc³V, Ga, Ge) pairs with large atomic sizes, although it does not change the sign of the interaction energies (distinction between repulsion and attraction). The large relaxation for ISn pairs may be mainly due to the large atomic sizes of the impurities compared to that of the host atom, as discussed in Sect. 4.
It is important to explain why the lattice distortion effect on ISn (I=Co³Cu) interaction energy becomes small, although the lattice distortion caused by a Sn impurity is very high (expansion of 3.4% and lattice distortion energy of ¹0.538 eV), as shown in Fig. 10. Based on a chemical bond picture, the interaction energy of ISn is defined by the total-energy difference due to the atomic rearrangement where the
Fig. 8 Distance dependence (1st10th, solid line) of interaction energies of ferromagnetic coupling MnMn (↓↓) to the bulk magnetization of Fe (↑). The solutions (broken line) of the antiferromagnetic coupling MnMn
(↑↓) for the 1st- and 2nd-NN distances are also shown. Fig. 9in Fe, including the lattice distortion effect, and the available experimentalCalculated interaction energies of 1st-NN ISn (I=Sc³Ge) pairs results.14) The calculated results for the 1st- and 2nd-NN interaction
[image:7.595.292.540.104.382.2]energies without the lattice distortion effect are also shown for comparison.
Table 3 Calculated interaction energies of the 1st-NN ISn (I=Sc³Ge) pairs in Fe with and without the lattice distortion effect, and the available experimental PAC results (in eV),14)and the calculated results for the 2nd-NN ISn interaction energies without the lattice
distortion effect. The changes in the ISn interatomic distances (in parentheses) are also listed (in%).
Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge
1st
Without
distortion 0.40 0.39 0.32 0.25 0.03 0.00 ¹0.04 ¹0.19 ¹0.19 0.08 0.38 0.63
With distortion
0.17 (8.9)
0.20 (8.5)
0.16 (7.6)
0.12 (7.7)
¹0.03 (7.0)
0.0 (3.4)
¹0.04 (3.5)
¹0.16 (3.4)
¹0.15 (3.5)
0.06 (5.5)
0.29 (9.6)
0.43 (10.5)
2nd Without
distortion 0.11 0.10 0.11 0.14 0.14 0.00 0.05 0.08 0.02 0.09 0.17 0.23
[image:7.595.45.549.104.207.2] [image:7.595.74.262.238.355.2] [image:7.595.310.542.240.380.2]two bonds (FeSn and IFe) are broken and the two bonds (ISn and FeFe) are created.21)Among the four bonds, the
lattice distortion is very low for IFe (I=Mn³Cu) bonds as shown in Fig. 3 and Table 1 because the atomic sizes of impurities I are almost the same as that of the Fe host atom, shown in Fig. 5. The lattice distortion energies of ISn (I=Co³Cu) bonds are similar to that of FeSn (¹0.54 eV) bond due to the same reason for IFe and FeFe bonds. For example, the lattice distortion energy for CoSn bond is as high as ¹0.528 eV, being very close to that for FeSn (¹0.538 eV), while it is as low as ¹0.001 eV for CoFe, being very close to that of FeFe (0 eV; no lattice distortion). Thus, the small lattice distortion effect on ISn (I=Co³ Cu) interaction energies may be understood by considering that the lattice distortion effect for the ISn bonds is cancelled out by that for FeSn bond.
We now discuss the magnetism of ISn pair. Figure 11 shows the calculated results for the magnetic moments of the ISn (I=Sc³Ge) pairs in Fe. For comparison, we show the magnetic moments of single impurities in Fe. Wefind that the magnetic moments of the Sn and I impurities do not change very much by the pairing of ISn, except for CrSn and Mn Sn pairs. It is noted that the lattice distortion (the change in interatomic distance) is high for CrSn (7.7%) and MnSn (7.0%) pairs (see Table 2), and that the magnetic moment of impurity I (Cr, Mn) changes from (¹1.52 and ¹1.61 µB) to
(¹1.61 and¹1.88 µB) by the displacement (5.4 and 5.4%) of
impurity I (Cr, Mn) from the ideal position towards the 1st-NN Fe atom on the opposite site of a Sn impurity. Increasing
the negative magnetic moment of the impurity obviously indicates that the antiferromagnetic coupling of FeI (I=Cr, Mn) becomes strong. Thus we may conclude that the large displacement of impurity I (Cr, Mn) is partly caused by the antiferromagnetic interaction of FeI (I=Cr, Mn) pairs, resulting in the significant energy gain (0.13 and 0.06 eV) of ISn (I=Cr, Mn) interaction energies, as shown in Fig. 9.
6. Summary and Discussion
We systematically and quantitatively studied the lattice distortion effect of substitutional single impurities I (I= Sc³Ge, Sn) and substitutional two impurities, II and ISn, in bcc-Fe.
For the single impurities, we showed the calculated results for the lattice distortion around the impurities, the atomic volume changes per impurity, and magnetism of the impu-rities. The available experimental results for the lattice relaxation around the impurities and the relative atomic volume changes per impurity are reproduced very well by the present ab-initio calculations. We found that the chemical trend for the lattice distortion around the impurities I is almost the same as that of the calculated lattice parameters of metals I in the bcc structure. However, it is noted that the large expansion caused by a Zn impurity, compared to those caused by Ga and Ge impurities, is understood by considering the HF forces on the 5th-NN’s. We also showed that the magnetism of single impurities in bcc-Fe is understood by considering the 3d electron numbers per atom.21,2932)
[image:8.595.77.262.65.358.2]For II and ISn (I=Sc³Ge, Sn=PAC-probe) pairs, we found that the lattice distortion effect on the interaction energies becomes significant for two impurities with a large size-misfit compared to the host atom, but very small for two impurities with a small size-misfit. The lattice distortion effect is lower than 0.02 eV for II (I=Cr³Zn). For ISn (I=Mn³Zn) pairs, it becomes slightly higher although it is less than 0.05 eV. According to the present calculations, the main part of the large lattice distortion energies of ISn (I=Co³Cu) pairs is cancelled out by that of FeSn pair. As a result, the lattice distortion effect on ISn (I=Co³Cu)
[image:8.595.306.550.71.220.2]Fig. 10 Lattice distortion energy (lower panel, parabolic behavior), radial force (lower panel, line) on the 1st-NN host atom, and magnetic moments (upper panel, two lines) of a Sn impurity and those of the 1st-NN host atom, as a function of the change of the 1st-NN distance. The magnetic moments of Sn (¹0.08 µB) and 1st-Fe (2.16 µB) don’t change very much.
interaction energies becomes low. We also found that we can improve the agreement with the PAC experimental results for the ISn (I=Co³Cu) interaction energies by taking into account the lattice distortion.
For the magnetism of II pairs, we found that the magnetic moments of the single impurities (I=Sc³Ge) do not change very much by the pairing of II, except Mn. For a MnMn pair in bcc-Fe, there are two solutions for magnet-ism, namely (↑↑) and (↑↓), to the bulk magnetization of Fe (↑). The antiferromagnetic interaction of the 1st-NN MnMn pair is very strong compared to the antiferromagnetic interaction of the FeMn pair. As a result, the (↑↓) state of the MnMn pair is stable by 0.076 eV, compared to the (↓↓) state of the MnMn pair. We also found that the magnetic moments of II and ISn pairs do not change very much by taking into account the lattice distortion, except for ISn (I=Cr, Mn) pairs. It is noted that the large lattice distortion of ISn (I=Cr, Mn) pairs is partly caused by the strong antiferromagnetic interaction of impurities I with the 1st-NN host atom on the opposite site of an Sn impurity, resulting in the significant energy gain of ISn interaction energies. The present results may be important data to construct an accurate model potential for Molecular Dynamics, which are signifi -cantly required to study the physical mechanism of the stability of metastable and non-equilibrium structures of complex metals such as quasicrystals and bulk metallic glasses.33,34)
Acknowledgement
One of the authors (T. Hoshino) thanks Prof. H. Numakura for the useful discussions regarding the experimental data. The authors are grateful for the financial support from the Ministry of Education, Culture, Sports, Science and Tech-nology (JSPS KAKENHI Grant Nos. 23560792 and 24560801).
REFERENCES
1) G. Liebfried and N. Breuer: Point Defects in Metals I, (Springer, Verlag, Berlin, 1978).
2) Y. Kraftmakher: Equilibrium Point Defects and Thermophysical Properties of Metals, (World Scientific, 2000).
3) F. R. de Bore, R. Boom, W. C. M. Mattens, A. Miedema and A. K. Niessen:Cohesion in Metals, (North-Holland, Amsterdam, 1988). 4) D. de. Fontaine: Solid State Physics, Vol. 47, ed. by H. Ehrenreich and
D. Turnbull, (Academic Press, London, 1994) pp. 33176.
5) O. I. Gorbatov, P. A. Korzhavyi, A. V. Ruban, B. Johansson and Yu. N. Gornostyrev:J. Nucl. Matter.419(2011) 248255.
6) G. Rahman, I. G. Kim, H. K. D. H. Bhadeshia and A. J. Freeman:Phys. Rev. B81(2010) 184423.
7) T. Tsuru, C. Suzuki, Y. Kaji and T. Tsukada:J. Appl. Phys.107(2010)
061805.
8) T. Hoshino, T. Mizuno, M. Asato and H. Fukushima:Mater. Trans.42
(2001) 22062215.
9) M. Asato, T. Mizuno, T. Hoshino and H. Sawada:Mater. Trans.42
(2001) 22162224.
10) T. Hoshino, W. Schweika, R. Zeller and P. H. Dederichs:Phys. Rev. B
47(1993) 5106.
11) T. Hoshino, M. Asato, R. Zeller and P. H. Dederichs:Phys. Rev. B70
(2004) 094118.
12) M. Asato, M. Ohkubo, T. Hoshino, F. Nakamura, N. Fujima and T. Tatsuoka:Mater. Trans.49(2008) 17601767.
13) T. Hoshino, N. Fujima, M. Asato and H. Tatsuoka:J. Alloy. Compd.
504S(2010) S531S533.
14) K. Królas:Hyperfine Interact.60(1990) 581597.
15) T. Hoshino, B. Drittler, R. Zeller and P. H. Dederichs:Phys. Rev. B45
(1992) 12202.
16) T. Hoshino, A. Shimizu, R. Zeller and P. H. Dederichs:Phys. Rev. B53
(1996) 5247.
17) T. B. Massalski, H. Okamoto, P. R. Subramabian and L. Kacprazak (ed.):Binary Alloy Phase Diagrams2nd ed., (ASM International, New York, 1990).
18) T. Hoshino, M. Asato and N. Fujima:Intermetallics14(2006) 908 912.
19) T. Hoshino, M. Asato, S. Tanaka, F. Nakamura and N. Fujima:
Intermetallics14(2006) 913916.
20) T. Hoshino, N. Fujima and M. Asato:J. Alloy. Compd.504S(2010) S534S537.
21) C. Liu, M. Asato, N. Fujima and T. Hoshino:Mater. Trans.54(2013) 1667.
22) N. Papanikolaou, R. Zeller, P. H. Dederichs and N. Stefanou: Phys. Rev. B55(1997) 4157.
23) T. Hoshino, N. Papanikolaou, R. Zeller, P. H. Dederichs, M. Asato, T. Asada and N. Stefanou:Comput. Mater. Sci.14(1999) 56.
24) C. Liu, M. Asato, N. Fujima and T. Hoshino: Proc. 8th Pacific Rim Int. Congress on Advanced Materials and Processing, (2013) pp. 2821 2825.
25) H. W. King:J. Mater. Sci.1(1966) 79.
26) U. Scheuer and B. Lengler:Phys. Rev. B44(1991) 9883.
27) We have already calculated the distance dependence of the HF forces for 3d-impurities in Al, which seems to be the simple oscillation mainly due to the screening effect of free-electron-like 3sp-electrons.28)For the
distance dependence of impurity-impurity interaction energies, please compare Fig. 3 in Ref. 21) with Fig. 3 in Ref. 19). The complicated behavior for 3d-impurities in Fe may be due to the interaction of dd interaction of IFe (I=3d impurities), in addition to the spd interaction of IFe.
28) C. Liu, M. Asato, N. Fujima and T. Hoshino: The 15th-IUMRS-International Conference in Asia submitted.
29) T. Moriya:Prog. Theor. Phys.33(1965) 157.
30) P. H. Dederichs, H. Akai, S. Blugel, N. Stefanou and. R. Zeller:Alloy Phase Stability, Series E, Applied Sciences, V. 163, (1989) pp. 377 420.
31) T. Hoshino, R. Zeller, P. H. Dederichs and T. Asada:J. Magn. Magn. Mater.156/158(1996) 717.
32) T. Hoshino, R. Zeller and P. H. Dederichs:J. Magn. Magn. Mater.140 144(1995) 113.
33) M. Asato, R. Tamura, N. Fujima and T. Hoshino:Mater. Sci. V.561 565(2007) 12591262.