organic papers
o2490
Mendliket al. C8H13NO4 doi:10.1107/S160053680601840X Acta Cryst.(2006). E62, o2490–o2492
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
Methyl 3-amino-3-
N
,4-
O
-carbonyl-2,3,6-trideoxy-
a
-
L-
lyxo
-hexopyranoside
Matthew T. Mendlik,aRobert S. Coleman,aGuizhong Qi,b Todd L. Lowaryb‡ and Robert McDonaldb*§
aDepartment of Chemistry, The Ohio State
University, 100 West 18th Avenue, Columbus, Ohio 43210, USA, andbChemistry Department, University of Alberta, Edmonton, Alberta, Canada T6G 2G2
‡ At the Alberta Ingenuity Centre for Carbohydrate Science
§ At the X-ray Crystallography Laboratory
Correspondence e-mail: bob.mcdonald@ualberta.ca
Key indicators
Single-crystal X-ray study
T= 193 K
Mean(C–C) = 0.003 A˚
Rfactor = 0.035
wRfactor = 0.079 Data-to-parameter ratio = 9.4
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 4 May 2006 Accepted 17 May 2006
#2006 International Union of Crystallography
All rights reserved
The structure of the title compound, C8H13NO4, shows the
oxazolidone ring to be essentially planar, which forces the pyranoside ring into a2SOskew-boat conformation. The CH3
and OCH3groups both adopt pseudo-equatorial orientations.
Comment
The title compound, (I), is an intermediate in a novel route (Mendlik et al., 2006) for the preparation of glycosides of daunosamine, a sugar residue present in a number of natural products, e.g. the anticancer agent daunorubicin (II) (Arca-mone & Cassinelli, 1998). Compound (I) was obtained in excellent yield (95%) upon treatment of a solution of the aziridine-containing methyl glycoside (III) in ethyl acetate with hydrogen and palladium on carbon. The presence of the oxazolidone ring in (I) altered the conformation of the pyranose ring from the usual 4C1 chair conformer, thus
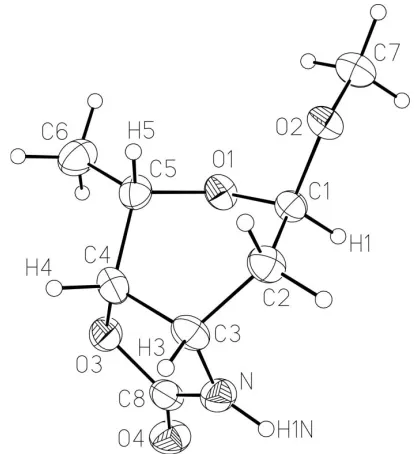
complicating the unequivocal determination of its structure by NMR spectroscopy. Fortunately, (I) could be crystallized and the X-ray structure analysis confirmed the expected structure (Fig. 1).
The solid-state structure of (I) shows the oxazolidone ring to be nearly planar [N—C3—C4—O3 = 6.3 (2); atoms O3, O4, N, C3, C4, and C8 are at most 0.0481 (14) A˚ out of the least-squares plane]. The pyranose ring is thus forced into an unusual2SOskew-boat conformation. As defined by Berceset
al.(2001), the polar coordinates for the pyranoside ring in (I) are d= 1.13,’= 259, = 81. In this pyranose ring confor-mation, the methyl group attached to C5 adopts the most sterically favourable pseudo-equatorial position. The OCH3
group attached to the anomeric centre (C1) is also in the pseudo-equatorial orientation. The placement of the methoxy group pseudo-equatorially is not only favoured for steric reasons, but this orientation also allows the molecule to be stabilized by the endo-anomeric effect (Lemieux & Koto, 1974). The orientation about the C1—O2 bond is the one favoured by theexo-anomeric effect (Lemieux & Koto, 1974),
The methoxy O atom is an acceptor of a proton from the
oxazolidone NH group of an adjacent molecule
(Fig. 2), generating a hydrogen-bonded helix running parallel to thebaxis.
Experimental
Methyl 2,3-diamino-3-N-,4-O-carbonyl-2,3-N-cyclo-2,3,6-trideoxy- -l-talopyranoside, (III) (460 mg, 2.48 mmol), and 10% palladium on
carbon (400 mg, 0.62 mmol Pd) were dissolved in EtOAc (20 ml). The reaction vessel was charged with a balloon of H2gas and the reaction
mixture was stirred at room temperature for 3 h, when it was filtered through Celite. The filtrate was concentrated and the residue was filtered through silica gel (1 3 cm, 5:1 chloroform/methanol) to afford (I) as white crystalline plates (440 mg, 95%). The solid was recrystallized from 3:1 EtOAc–hexane (m.p. 390–393 K).RF0.49 (6:1,
chloroform/methanol); []D
23
47.1 (c0.4, CHCl3); IR: 2936, 2253,
1757 cm1; 1H NMR (400 MHz, CDCl
3): 6.87 (s, 1H, NH), 4.84
(app. t, 1H,J= 6.0 Hz, C1—H), 4.46 (dd, 1H,J= 1.7, 8.8 Hz, C4—H), 4.19 (app. dt, 1H,J= 4.3, 8.8 Hz, C3—H), 3.99 (dq, 1H,J= 1.7, 6.6 Hz, C5—H), 2.08 (app. dt, 1H,J= 4.9, 15.0 Hz, C2—Hb), 1.75 (ddd, 1H,
J= 4.3, 6.0, 15.0 Hz, C2—H), 1.31 (d, 3H,J= 6.6 Hz C6—H);13C
NMR (100 MHz, CDCl3):160.0 (C O), 96.8 (C-1), 76.2 (C-4), 63.3
(C-5), 54.8 (OCH3), 47.8 (C-3), 30.0 (C-2), 15.6 (C-6); HRMS (ESI)
m/zcalculated for [C8H13NO4]Na
+: 210.0742, found: 210.0736.
Crystal data
C8H13NO4
Mr= 187.19
Orthorhombic,P212121
a= 5.2411 (8) A˚
b= 10.2928 (16) A˚
c= 16.760 (3) A˚
V= 904.1 (2) A˚3
Z= 4
Dx= 1.375 Mg m
3 MoKradiation
= 0.11 mm1
T= 193 (2) K Rod, colourless 0.590.120.10 mm
Data collection
Bruker SMART 1000 CCD area detector/PLATFORM diffractometer
!scans
Absorption correction: multi-scan (SADABS; Bruker, 2003)
Tmin= 0.739,Tmax= 0.989
6168 measured reflections 1114 independent reflections 940 reflections withI> 2(I)
Rint= 0.052
max= 26.4
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.035
wR(F2) = 0.079
S= 1.04 1114 reflections 118 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0377P)2 + 0.1378P]
whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.15 e A˚
3 min=0.18 e A˚
[image:2.610.62.269.74.301.2]3
Table 1
Selected geometric parameters (A˚ ,).
O1—C1 1.410 (3) O1—C5 1.435 (3) O2—C1 1.424 (2) O2—C7 1.425 (3) O3—C4 1.447 (3) O3—C8 1.366 (3) O4—C8 1.212 (3)
N—C3 1.454 (3) N—C8 1.335 (3) C1—C2 1.518 (3) C2—C3 1.522 (3) C3—C4 1.545 (3) C4—C5 1.508 (3) C5—C6 1.508 (3)
C1—O1—C5 114.36 (16) C1—O2—C7 112.79 (17) C4—O3—C8 110.17 (17) C3—N—C8 113.8 (2) O1—C1—O2 110.95 (18) O1—C1—C2 113.31 (18) O2—C1—C2 105.41 (17) C1—C2—C3 112.93 (19) N—C3—C2 113.69 (19) N—C3—C4 100.78 (18)
C2—C3—C4 112.03 (19) O3—C4—C3 105.32 (17) O3—C4—C5 109.24 (18) C3—C4—C5 111.81 (18) O1—C5—C4 108.35 (18) O1—C5—C6 108.11 (19) C4—C5—C6 113.75 (19) O3—C8—N 109.4 (2) O4—C8—N 129.3 (2) O3—C8—O4 121.3 (2)
organic papers
Acta Cryst.(2006). E62, o2490–o2492 Mendliket al. C
[image:2.610.95.235.354.578.2]8H13NO4
o2491
Figure 1
The molecular structure of (I), with displacement ellipsoids drawn at the 50% probability level.
Figure 2
Molecules are connected by N—H O hydrogen bonds (dotted lines),
creating a chain parallel to thebaxis along the 21screw operation (x,12+
[image:2.610.313.566.562.724.2]Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
N—H1N O2i
0.88 2.18 2.976 (3) 150
Symmetry code: (i)x;yþ1 2;zþ
1 2.
In the absence of significant anomalous dispersion effects, Freidel pairs were merged. The assignment of the absolute stereochemistry is based on the known stereochemistries of the optically pure precursor compounds. H atoms were placed in idealized positions (N—H = 0.88 A˚ and C—H = 0.98–1.00 A˚) and these were assigned isotropic displacement parameters 1.2 timesUeqof their parent C atoms.
Data collection:SMART(Bruker, 2001); cell refinement:SAINT
(Bruker, 2003); data reduction: SAINT; program(s) used to solve structure: SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
SHELXTL (Bruker, 2003); software used to prepare material for publication:SHELXTL.
This work was supported by the Natural Sciences and Engineering Research Council of Canada, the Alberta Inge-nuity Centre for Carbohydrate Science, and the National Institutes of Health.
References
Arcamone, F. & Cassinelli, G. (1998).Curr. Med. Chem.5, 391–419. Berces, A., Nukada, T. & Whitfield, D. M. (2001).Tetrahedron,57, 477–491. Bruker (2001).SMART. Version 5.054. Bruker AXS Inc., Madison, Wisconsin,
USA.
Bruker (2003). SADABS (Version 2.10), SAINT (Version 7.06A) and
SHELXTL(Version 6.14). Bruker AXS Inc., Madison, Wisconsin, USA. Lemieux, R. U. & Koto, S. (1974).Tetrahedron,30, 1933–1944.
Mendlik, M. T., Tao, P., Hadad, C. M., Coleman, R. S. & Lowary, T. L. (2006).J. Org. Chem.Submitted.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
organic papers
o2492
Mendliket al. Csupporting information
sup-1
Acta Cryst. (2006). E62, o2490–o2492
supporting information
Acta Cryst. (2006). E62, o2490–o2492 [https://doi.org/10.1107/S160053680601840X]
Methyl 3-amino-3-
N
,4-
O
-carbonyl-2,3,6-trideoxy-
α
-
L-
lyxo
-hexopyranoside
Matthew T. Mendlik, Robert S. Coleman, Guizhong Qi, Todd L. Lowary and Robert McDonald
methyl 3-amino-3-N,4-O-carbonyl-2,3,6-trideoxy-α-L-lyxo-hexopyranoside
Crystal data
C8H13NO4
Mr = 187.19
Orthorhombic, P212121
Hall symbol: P 2ac 2ab
a = 5.2411 (8) Å
b = 10.2928 (16) Å
c = 16.760 (3) Å
V = 904.1 (2) Å3
Z = 4
F(000) = 400
Dx = 1.375 Mg m−3
Melting point: 390 K
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 1719 reflections
θ = 2.3–23.7°
µ = 0.11 mm−1
T = 193 K Rod, colourless 0.59 × 0.12 × 0.10 mm
Data collection
Bruker SMART 1000 CCD area detector/PLATFORM diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 8.192 pixels mm-1
ω scans
Absorption correction: multi-scan (SADABS; Bruker, 2003)
Tmin = 0.739, Tmax = 0.989
6168 measured reflections 1114 independent reflections 940 reflections with I > 2σ(I)
Rint = 0.052
θmax = 26.4°, θmin = 2.3°
h = −6→6
k = −12→12
l = −18→20
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.035
wR(F2) = 0.079
S = 1.04 1114 reflections 118 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0377P)2 + 0.1378P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.15 e Å−3
Δρmin = −0.18 e Å−3
Special details
supporting information
sup-2
Acta Cryst. (2006). E62, o2490–o2492
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.3424 (3) 0.43448 (14) 0.39952 (9) 0.0281 (4) O2 0.1518 (3) 0.25412 (13) 0.33959 (9) 0.0283 (4) O3 0.2438 (3) 0.70392 (14) 0.43647 (9) 0.0319 (4) O4 0.3947 (4) 0.83464 (15) 0.33993 (10) 0.0396 (4) N 0.0851 (4) 0.68113 (17) 0.31545 (12) 0.0319 (5) H1N 0.0704 0.6910 0.2635 0.038* C1 0.1977 (4) 0.3902 (2) 0.33401 (14) 0.0257 (5) H1 0.2935 0.4085 0.2837 0.031* C2 −0.0644 (5) 0.4523 (2) 0.32874 (14) 0.0296 (5) H2A −0.1876 0.3987 0.3591 0.036* H2B −0.1199 0.4533 0.2723 0.036* C3 −0.0697 (5) 0.5905 (2) 0.36109 (13) 0.0287 (5) H3 −0.2497 0.6224 0.3641 0.034* C4 0.0582 (5) 0.6006 (2) 0.44389 (13) 0.0281 (5) H4 −0.0716 0.6235 0.4853 0.034* C5 0.1930 (5) 0.4765 (2) 0.46646 (14) 0.0280 (5) H5 0.0623 0.4085 0.4786 0.034* C6 0.3684 (5) 0.4903 (2) 0.53725 (16) 0.0396 (6) H6A 0.4486 0.4064 0.5488 0.047* H6B 0.2699 0.5189 0.5838 0.047* H6C 0.5006 0.5547 0.5252 0.047* C7 0.3807 (5) 0.1794 (2) 0.33806 (15) 0.0370 (6) H7A 0.3388 0.0870 0.3430 0.044* H7B 0.4906 0.2055 0.3826 0.044* H7C 0.4702 0.1945 0.2876 0.044* C8 0.2515 (5) 0.7480 (2) 0.35963 (14) 0.0292 (5)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3
Acta Cryst. (2006). E62, o2490–o2492
C6 0.0475 (15) 0.0390 (14) 0.0321 (13) −0.0075 (14) −0.0072 (14) 0.0039 (11) C7 0.0397 (15) 0.0267 (12) 0.0445 (15) 0.0056 (11) 0.0004 (14) −0.0002 (11) C8 0.0313 (12) 0.0230 (11) 0.0333 (13) 0.0036 (11) −0.0027 (11) −0.0018 (10)
Geometric parameters (Å, º)
O1—C1 1.410 (3) C2—H2B 0.9900 O1—C5 1.435 (3) C3—C4 1.545 (3) O2—C1 1.424 (2) C3—H3 1.0000 O2—C7 1.425 (3) C4—C5 1.508 (3) O3—C4 1.447 (3) C4—H4 1.0000 O3—C8 1.366 (3) C5—C6 1.508 (3) O4—C8 1.212 (3) C5—H5 1.0000 N—C3 1.454 (3) C6—H6A 0.9800 N—C8 1.335 (3) C6—H6B 0.9800 N—H1N 0.8800 C6—H6C 0.9800 C1—C2 1.518 (3) C7—H7A 0.9800 C1—H1 1.0000 C7—H7B 0.9800 C2—C3 1.522 (3) C7—H7C 0.9800 C2—H2A 0.9900
supporting information
sup-4
Acta Cryst. (2006). E62, o2490–o2492
C5—O1—C1—O2 −90.4 (2) C3—N—C8—O4 −175.4 (2) C5—O1—C1—C2 28.0 (3) O1—C1—C2—C3 31.2 (3) C1—O1—C5—C4 −69.5 (2) O2—C1—C2—C3 152.66 (18) C1—O1—C5—C6 166.80 (17) C1—C2—C3—N 64.7 (3) C7—O2—C1—O1 −62.1 (2) C1—C2—C3—C4 −48.8 (3) C7—O2—C1—C2 174.87 (18) N—C3—C4—O3 6.3 (2) C8—O3—C4—C3 −4.1 (2) N—C3—C4—C5 −112.2 (2) C8—O3—C4—C5 116.1 (2) C2—C3—C4—O3 127.5 (2) C4—O3—C8—O4 −179.9 (2) C2—C3—C4—C5 9.0 (3) C4—O3—C8—N −0.2 (3) O3—C4—C5—O1 −69.3 (2) C8—N—C3—C2 −127.1 (2) O3—C4—C5—C6 50.9 (3) C8—N—C3—C4 −7.0 (3) C3—C4—C5—O1 46.9 (2) C3—N—C8—O3 4.9 (3) C3—C4—C5—C6 167.1 (2)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N—H1N···O2i 0.88 2.18 2.976 (3) 150