IMPROVING SIGNAL IDENTIFICATION FOR FAST-SCAN CYCLIC VOLTAMMETRY
163
0
0
Full text
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
Figure



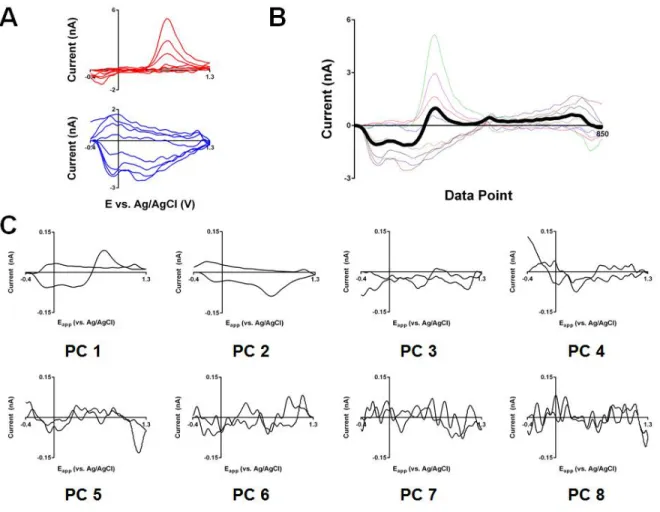
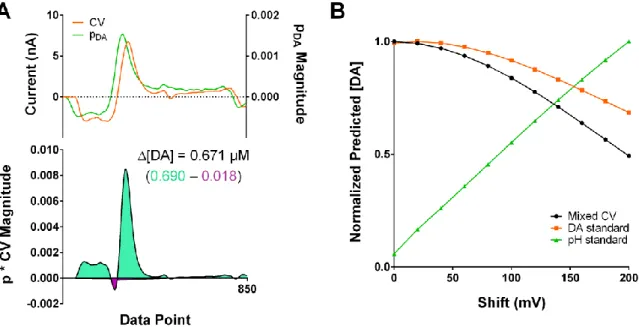
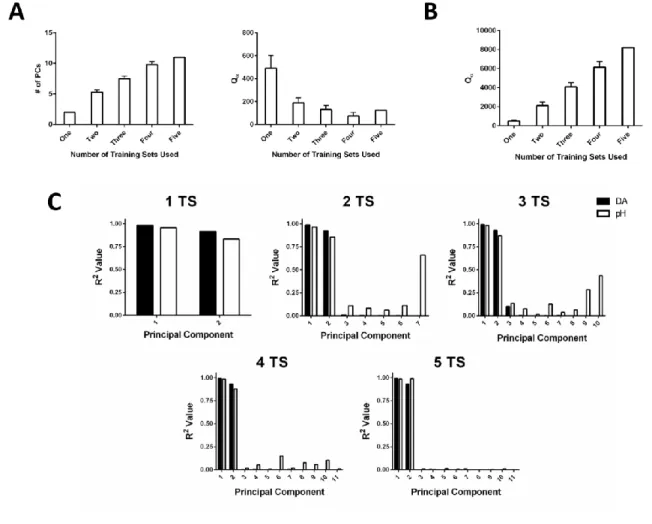
+7




Related documents
• When there is a reasonable selection of information interpreted under an incorrect top level frame (e.g. air travel vs ground travel) it provides a correct top
equipped for real-time visualization of the results shows that the continuous infusion of insulin (between 0.02 U/ kg/h and 0.06 U/kg/h), guided by glucose levels, enabled us to
EURASIP Journal on Wireless Communications and Networking 2005 5, 645?660 c? 2005 Bosheng Zhou et al A Cross Layer Route Discovery Framework for Mobile Ad Hoc Networks Bosheng Zhou
7 [18] we compare the measurement of the nuclear modification factor for inclusive primary charged-particle ( h ± ) production in p–Pb collisions to that in central (0–5 %
The unsteady flow with head transfer of a dusty couple stress conducting fluid under the influence of an applied uniform magnetic field has been studied in the presence of
This article in particular explores professional communi- cation represented in modern-day popular science publica- tions on the topic of information technologies. The choice of
All measures soil properties in this study could explain higher significant variability of the response variables (SOC, TN, SMBC and SMBN contents), suggesting
Key points: 1) Clearly, a significant amount of fructose can be converted to lactate, but quantitative metabolic data of dietary fructose to lactate conversion is very lim- ited.
