Combining Classical Molecular Dynamics and Quantum
Mechanical Methods for the Description of Electronic
Excitations: The Case of Carotenoids
Ingrid G. Prandi,
[a]Lucas Viani,
[a,b]Oliviero Andreussi,
[a]and Benedetta Mennucci*
[a]Carotenoids are important actors both in light-harvesting (LH) and in photoprotection functions of photosynthetic pigment– protein complexes. A deep theoretical investigation of this multiple role is still missing owing to the difficulty of describ-ing the delicate interplay between electronic and nuclear degrees of freedom. A possible strategy is to combine accu-rate quantum mechanical (QM) methods with classical molecu-lar dynamics. To do this, however, accurate force–fields (FF) are necessary. This article presents a new FF for the different carotenoids present in LH complexes of plants. The results
show that all the important structural properties described by the new FF are in very good agreement with QM reference values. This increased accuracy in the simulation of the struc-tural fluctuations is also reflected in the description of excited states. Both the energy order and the different nature of the lowest singlet states are preserved during the dynamics when the new FF is used, whereas an unphysical mixing is found when a standard FF is used.VC 2016 Wiley Periodicals, Inc.
DOI: 10.1002/jcc.24286
Introduction
Carotenoids are naturally occurring pigments in different light-harvesting (LH) pigment–protein complexes (PPC) of photosyn-thetic organisms of plants, bacteria, and algae.[1,2] Apart from increasing the structural stability of the PPCs and enhancing their energy absorption by supporting the most abundant (bacterio)chlorophyll pigments, carotenoids also have a photo-protective role, regulating the flow of energy within the pho-tosynthetic apparatus and protecting it from the damage owing to an excess light absorption.[3–10]
The understanding of the different roles of carotenoids in LH systems was made possible mainly owing to the advances in X-ray diffraction techniques, now able to resolve the crystal-line structures of the complexes at a good level of accu-racy.[11–14] By combining these structural data with spectroscopic techniques, it was possible to interpret the com-plex cascade of processes that are initiated by the absorption of sunlight. In parallel, the detailed information on the struc-tural properties of the complexes has allowed the use of com-putational chemistry techniques to support the experimental findings, and to predict the properties that are not possible to be assessed using the current experimental techniques. A common computational strategy used in the study of PPCs is the combination of classical molecular dynamics (MD) simula-tions with quantum mechanical (QM) calculasimula-tions of the elec-tronic processes.[15–25] The MD simulations allow the study of the PPCs in different environments, as well as the realistic sim-ulation of thein vivoconditions, by inserting the complex into a solvated lipid membrane.[20–22]
Typically, the MD trajectories are applied to obtain structural information used in different analyses, such as the calculation of the spatial correlations between the atomic motions and the normal mode analysis.[23,24] At the same time, the
evolu-tion of the electronic properties in time provides direct access to excitonic properties such as site energies, excitonic, and exciton–phonon couplings, used as input parameters for exci-ton dynamic models.[16,18,25]It is evident that in all these anal-yses the quality of the molecular mechanics (MM) force–fields (FF) used to describe the protein backbone, the solvent, and the cofactors (including the pigments) plays a fundamental role. Although accurate FF optimized for proteins and solvents are nowadays available, nonspecific FF, also referred to as gen-eral FF (such as Amber Gengen-eral FF[26] and CHARMM General FF[27]), are commonly used in the description of the cofactors. Such general FF may be effectively applied if the cofactors are not directly involved in the process under study or if the pro-cess is not significantly affected by structural deformations. Unfortunately, this is not the case of LH processes where the absorption of light and the following excitation energy trans-fers are highly sensitive to small changes in the intra- and intermolecular arrangements of the pigments. In the case of carotenoids, the situation is more delicate as their excited
[a]I. G. Prandi, L. Viani, O. Andreussi, B. Mennucci
Dipartimento Di Chimica E Chimica Industriale, University of Pisa, via G. Moruzzi 13, Pisa, I-56124, Italy
E-mail: [email protected]
[b]L. Viani
Institute for Fluid Dynamics, Nanoscience and Industrial Mathematics, Universidad Carlos III De Madrid, Av. De La Universidad 30, Leganes, E-28911, Spain
Contract grant sponsor: The European Research Council (ERC); Contract grant number: 277755; Contract grant sponsor: CNPq—Brazil; Contract grant number: 236693/2012-3; Contract grant sponsor: The Universidad Carlos III de Madrid, The European Commission (7th Framework Programme); Contract grant number: 600371; Contract grant sponsor: el Ministerio de Economıa y Competitividad of Spain; Contract grant number: COFUND2013-40258; Contract grant sponsor: Banco Santander
states (those involved in the LH and photoprotection functions) are largely dependent on the structural properties.[1,28–37] In particular, the use of a general FF presents three main problems when applied to carotenoids: (i) the overestimation of the bond length alternation (BLA), making the single bonds longer and the double bonds shorter compared to those obtained at accu-rate QM levels; (ii) the underestimation of the torsion barriers for some dihedrals, which may lead to unphysical perturbations in the conjugation path and to cis/trans conformational changes; and (iii) an incorrect description of the normal modes involving the conjugated chain. All these effects are extremely negative when the MD simulation is used in combination with QM methodologies as the conformational space sampled in the MD simulations is not a good enough approximation to the QM one, and thus leading to incorrect description of the elec-tronic properties and their fluctuations.
Up to now, some proposals of specific FF for LH pigments have been presented for Amber and CHARMM FF,[38–40] but they have been mainly focused on the family of chlorophylls and/or not explicitly validated to reproduce electronic proper-ties. To the best of our knowledge, FF specifically developed
to be combined with QM calculations of electronic excitations have never been proposed. In this study, we propose a new set of FF parameters for the different carotenoids found in the LH complexes of photosystem II, optimized to accurately reproduce not only the structural and vibrational properties, but also the electronic transition properties (transition energies and transition dipole moments) as obtained at density func-tional theory (DFT) level.
All the investigated carotenoids belong to the xanthophyll family: the molecular structure of xanthophylls is similar to that of carotenes, but they contain oxygen atoms, whereas carotenes are purely hydrocarbons with no oxygen. Xantho-phylls contain their oxygen either as hydroxyl and/or as epox-ide group.
In particular, the new parameterization has been done for the three xanthophylls found in the LH complexes of PSII, namely neoxanthin (NEX), violaxanthin (VIO), and lutein (LUT), as well as for a fourth one that is obtained from VIO through the enzymatic removal of the two epoxy groups to create the so-called de-epoxidized xanthophyll known as zeaxanthin (ZEA) (Fig. 1).
In the next sections, it will be shown that the most impor-tant structural properties, such as backbone CC BLA and normal-mode distribution, are described in close agreement to the reference DFT values when adopting the FF developed in this study. It will also be shown that the improved evaluation of the structural fluctuations is reflected in an accurate description of the singlet excited states, which maintain the energy ordering and the different natures, as predicted by DFT, even during the MD.
We start by presenting a short summary of the computa-tional tools adopted in this study (Computacomputa-tional Details sec-tion), followed by the description of the novel molecule-specific FF parameters (Derivation of FF parameters section), using a procedure which combines DFT calculations of ground-state structures and Hessians with self-consistent refinements (e.g., symmetrization and rescaling of the parame-ters). Finally, in Results section, the results of the structural and vibrational properties obtained with the new FFs are pre-sented and commented, followed by the characterization of electronic excitations.
Computational Details
MD simulations were performed using the AMBER 12[41] simu-lation package. The carotenoids were simulated in vacuum (no periodic boundary conditions and infinite Coulomb cutoff ) with a timestep of 1 fs. After a preliminary minimization step, the structures were equilibrated for 1 ns at room temperature,
T5300 K, using a Langevin thermostat with a characteristic relaxation time of 1.0 ps. Production runs of 10 ns were then performed with the same setup.
DFT calculations were performed using the Gaussian 09[42] package. Geometry optimizations of the four carotenoids were performed using the hybrid B3LYP functional and the 6-31G(d,p) basis set. The same setup was adopted in the calcula-tion of the Hessian matrices, and energy profiles along the internal degrees of freedom; this basis set size is known to represent a very good compromise between computational time and quality for structural analyses. To have a more detailed description of the electronic density, a larger basis set, namely 6-311G(d,p), was used for the derivation of atomic charges, using the standard electrostatic potential fitting approach, still exploiting the hybrid B3LYP functional, followed by the Restrained ElectroStatic Potential (RESP)[43]procedure.
Electronic excitations were computed using the Tamm– Dancoff Approximation (TDA) with the double-zeta 6-311G(d) basis set and the meta-GGA functional Tao–Perdew–Staro-verov–Scuseria (TPSS).[44] Recently, some of the authors have,
in fact, shown that the combination of TDA with meta-GGA functionals (and in particular TPSS) gives a generally good description of the transition properties of the lowest singlet states of carotenoids of different lengths.[33]
Derivation of FF Parameters
Consistently with most existing FF developed to model organic molecules and biological systems, the selected form of
the FF features a combination of bonded and nonbonded interactions. The latter ones were described using the long-range Coulomb interactions modeled through nonpolarizable atomic charges, and the short-range van der Waals terms, described by the 12-6 Lennard-Jones two-body potential. Atomic charges were determined via the standard RESP approach, whereas Lennard-Jones parameters were assigned according to each atomic element and defined according to GAFF.
For the bonded interactions, the parameters for bonds, angles, and dihedrals were derived according to the AMBER FF formulation, defined as:
VTotal5Vbond1Vangle1Vdihe5
5Xkbðr2reqÞ21 X
kaðu2ueqÞ21
1XVn
2 ½11cosðnu2gÞ
(1)
withkb,ka, andVn being the force constants; r andhare the bond length and bond angle, respectively; req and heq are
bond and angle equilibrium distance; n is the periodicity, u
corresponds to dihedral angle, andcis the phase.[45]
The definition of the different atom types and the assign-ment of an atom type to a specific atom of the molecule is a fundamental part of the generation of the FF. In particular, lim-iting the number of atom types is often pursued with the aim of reducing the number of fitting parameters and improving the transferability of the potential: as a matter of fact, this is the approach of some of the most common FF, including GAFF, whose use is wide spread in the literature. On the other hand, a general FF, based on a reduced number of atom types, usually comes at the price of a reduced accuracy in the description of the specific structural and vibrational features of the systems under study. Although such a reduction in accu-racy is usually not a severe issue for ground-state properties and the dynamics of the systems, it may critically affect the simulation of electronic properties, which are much more sen-sitive to the underlying description of the system. For such applications, FF with specific atom types can be generated: a few tools have been proposed in the literature, which allow to automatically fit on first-principle data, a set of parameters, corresponding to a number of atom types which could be as large as the number of atoms in the system.[27,46,47] Although such an atom-specific approach has some evident advantages over a general FF, it also presents some possible drawbacks: being parameterized on a single configuration, usually corre-sponding to a minimum energy structure of the system, the atom-specific FF may poorly describe different conformations of the same system or its dynamics far from the equilibrium structure. In addition, further tunings of the FF to improve the consistency with other experimental or theoretical data is hin-dered by the large number of parameters.
enforcing equivalence among chemical groups and atoms linked by symmetry operations and/or conformational equilib-rium. Bonds and angles between similar atom types were clus-tered in groups, which allowed a simpler collective fine tuning of the force constants to reproduce the QM normal modes. Eventually, dihedral angle parameters were either explicitly fit-ted on QM potential energy scans or assigned according to GAFF and to some recent literature on conjugated systems.
The details of the specification of atom types and the final values of all the FF parameters are reported in the Supporting Information Material.
Bond and angle parameters
The initial values of force constants and equilibrium values for bonds and angles were computed using a local harmonic approximation to describe the potential energy surface (PES) of the QM-minimized geometries. Different approaches have been developed for the calculation of force constants based on the Hessian values. The most straightforward method relies on the eigenvalue analysis Hessian matrix,[48] providing the force constants for the different internal coordinates (ICs). This method has been implemented in the Force-Field Toolkit[46] together with more iterative schemes based on the direct comparison between the MM and the QM Hessians or in the energetic variations owing to small distortions along the ICs. In this study, we have used the eigenvalue analysis scheme available as a plugin named Paratool[46] for the VMD soft-ware,[49] already correcting for the electrostatic contributions, naturally included in the QM Hessians. Force constants were appropriately scaled by the fitting program to take into
account the constant factor between first-principle and experi-mental vibrational frequencies as reported in the Minnesota Database of Frequency Scale Factors for Electronic Model Chemistries.[49,50]
Starting from the initial procedure, a different atom type was assigned to each atom of the systems, with a total of 100 atom types for NEX and VIO and 98 for LUT and ZEA. To reduce the amount of parameters and to improve the transfer-ability of the FF to different conformations of the studied mol-ecules, equivalence of certain molecular groups was enforced and bond and angle force constants were set to the average values of the equivalent terms. In particular, we considered equivalent (for some examples, see Fig. 2) (i) atoms belonging to different methyl groups (in the same carotenoid); (ii) hydro-gen atoms connected to the same carbon atom; and (iii) atoms connected by the C2 symmetry operations in the high-est symmetry conformers of VIO and ZEA; for NEX and LUT, portions of the conjugated chain backbones with similar “isoprene-like” structure were also considered equivalent. Fol-lowing the symmetrization procedure, the numbers of atom types were reduced to 62, 45, 37, and 35 for NEX, LUT, VIO, and ZEA, respectively.
factor was determined for each group (Supporting Information Table S1). In particular, bonds were divided into eight groups and the homogeneous scaling factors were fitted in this order: methyl CAH, methyl CAC, ring CAH, ring CAC, backbone CAC, backbone C@C, backbone CAH, and OAH bonds. Simi-larly, angle force constants were divided into other eight groups and their homogeneous scaling factors were tuned in this order: methyl HACAH, methyl CACAH, ring CACAC, ring CACAH, ring HACAH, backbone CACAC, backbone CACAH, and finally, CACAO.
Dihedral force parameters
The allene in NEX and the backbone-ring dihedral constants (Supporting Information Fig. S1) were set to provide the best fit between the QM and the MM dihedral PES rigid scans. Dihedral parameters for the atoms of the conjugated chains of the carotenoids were assigned by following the recent study from Cerezo et al.[40] on extended conjugated systems. The remaining parameters were derived from GAFF[26] and homogeneously scaled to improve the agreement with the low-energy region of the simulated IR spectrum of the carotenoids.
Results
The newly developed FF was tested with respect to structural, vibrational, and electronic properties. MD simulations were car-ried out for each of the four carotenoids in gas phase and the resulting snapshots were used to evaluate the averaged prop-erties and fluctuations.
Structural properties
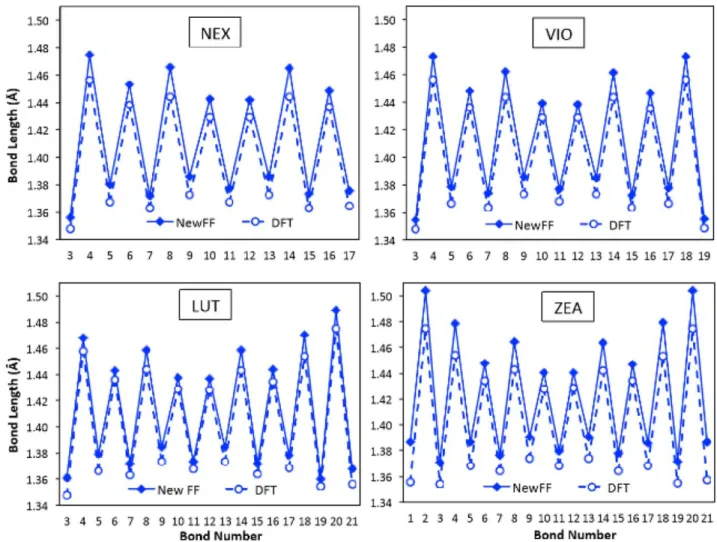
As discussed in the Introduction section, the degree of con-jugation of a carotenoid’s backbone is known to have a strong influence on its electronic properties as well as on the electronic excitations. From a structural point of view, the most straightforward measure of such a conjugation is represented by the elongation/contraction of adjacent single and double bonds. In Figure 3, the average bond lengths obtained from the MD simulations using the new FF and the optimized DFT results are shown for the four carotenoids considered in this study (the number that identifies each bond is shown in Fig. 1). All the DFT and MD bond lengths and the (MD) standard deviations are listed in Supporting Information Table S2.
The trend in bond lengths found in DFT calculations is well reproduced by the newly developed FF: all the carotenoids behave similarly, with more conjugated bonds at the center of the backbone, where more elongated (contracted) single (dou-ble) bonds are found at the borders. The main exception is represented by the two most external double bonds of ZEA that show an elongation with respect to, for example, the other double bonds of the same molecule and the structurally similar external double bond of LUT. It is evident that these specificities of the double (single) bonds related to their posi-tions along the backbone would be inaccessible to any gen-eral FF that rely on only two atom types to describe the conjugated carbon atoms.
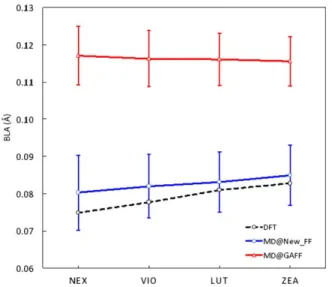
To verify the quality of the structural description of the backbone described with the new set of parameters, the BLA analysis was also performed (Fig. 4).
It has to be noted that there are deviations in BLA values among different carotenoids, even for those with similar back-bone structures. By comparing these results with the data shown in Figure 3, it is clear that the BLA differences are mostly owing to the lengths of terminal conjugated bonds, which present large deviations with respect to the central part of the backbone. Indeed, the conjugation in ZEA and LUT comprises the carotenoid’s rings and the terminal bond lengths that are more similar to “localized” systems, resulting in larger BLA values.
From the graph, it is evident that the new parameterization leads to an average picture that is in good agreement with the DFT results for all the systems (Supporting Information Table S3). In particular, both the correct order of magnitude of the BLA and the correct trend along the series of carotenoids are quantitatively reproduced by the new FF. On the contrary, the GAFF parameterization systematically leads to an overlocalized picture with double bonds being too short and single ones too long as well it misses the trends along the carotenoid series,
resulting in very similar BLA values for all four carotenoids as they are described by just two different atom types.
Normal mode analysis
To further validate the newly developed parameters (Fig. 5), we compare the normal mode distributions obtained through DFT and the new FF for the four carotenoids. The “count” in the y-axis describes the amount of different normal modes present in the frequency interval represented by the width of the histograms bars. The normal mode distributions were com-puted for the minimized structure of each carotenoid by the diagonalization of Hessian matrix.[45]
As it can be seen from the graphs, the agreement between DFT and MM distributions is very satisfactory for all carote-noids when the new parameters are used. The best correspon-dence is observed in the mid-high-frequency region Figure 4. BLA of the four carotenoids computed from the DFT-optimized
structures (dotted black line) and as averages along the classical MD simu-lations using either GAFF (red line) or the new (blue line) FF. Error bars reported for MD results refer to the standard deviation. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
dominated by modes related to angle and bond stretching. At the low-frequency region, only the modes connected to the backbone were refined to improve the representation of the modes playing a significant role in the prediction of excited state properties.
Together with bringing the normal modes in the correct fre-quency region, the rescaling of the force constants allowed to preserve the correct ordering of the main vibrational modes, yielding reliable FF when used in vibrational analysis. In Figure 6, we show a comparison between the atomic displacement vectors of the modes playing an important role in the descrip-tion of excited state properties of LUT, as computed in the QM and MM framework.
As it can be seen, the DFT description of the selected nor-mal modes is correctly reproduced by the classical description, with small differences probably reflecting the enforcement of equivalence between atom types in the MM description. In addition, the mean percentage error between QM and MM fre-quencies for these modes is <2%, further validating the good agreement between the two sets of results. A similar agree-ment is also found for the other three carotenoids as shown in Supporting Information Figure S2.
Excitation properties
It is common to represent the nature of the carotenoids’ elec-tronically excited states using an ideal picture of ap-system in a perfect C2h symmetry; according to this model, the lowest
lying singlet state (S1) is described by the same irreducible representation as the ground state A2
g, whereas the second singlet excited state (S2) has B1u symmetry. An additional dark
B2
u state, termed S3, is also predicted by the model. This analy-sis implies that the S0 !S1 transition is (electric dipole) for-bidden and the lowest state accessible through light absorption is S2. The S1 state can thus be populated mainly by fast internal conversion from S2. Unfortunately, a detailed description of the electronic states of long carotenoids with QM approaches is still a challenge owing to the multireference
character of some of these states combined with the size of the systems, which prevents the use of very accurate and highly correlated methods. It is known that the various excited states have a rather different multireference character,[34–36,51] S2 being largely dominated by a single excitation, whereas S1 and S3 present strong double excitation characters, possibly mixed with single excitations.
As a result, single-reference approaches only properly describe the S2 state, predicted as the lowest singlet state, whereas the correct energy position and character of, for example, the real S1 state is not achievable. Recently, some of the present authors have shown that the combination of TDA with meta-GGA functionals (and in particular TPSS) gives a generally good description of the energy ordering of the low-est singlet states of carotenoids of different lengths when compared to a multireference configuration–interaction exten-sion of DFT (DFT/MRCI).[33] Such analysis confirmed that this behavior is owing to the cancellation of the effects that the neglect of the multideterminant character has on the ground and the excited states (this also explains the reason why TDA gives a better picture than time-dependent DFT). From those results, it also appeared that this cancellation of errors does apply not only to excitation energies but also to transition densities (and the resulting transition dipoles). Following those findings, we have here applied the TDA/TPSS analysis to the three lowest singlet excited states of the four selected carote-noids along the MD trajectories, obtained either using the new FF or the GAFF.
Figures 7 and 8 show the corresponding transition energies as obtained from a set of 1000 noncorrelated geometries taken from the MD trajectory every 10 ps. Supporting Informa-tion Table S4 compares the results obtained on the DFT struc-tures and the MD average values and summarizes the spreads of the new FF and GAFF distributions for NEX.
the overlap between the first and the second transition ener-gies is reduced with respect to the GAFF results: in the partic-ular case of NEX, mean oscillation amplitudes of about 0.77 eV are obtained with GAFF, in contrast with the value of 0.39 eV, resulting from the new FF. These features suggest that the new FF describes the first three excited states in a more physi-cally stated way.
The correlation between the oscillation of the excitation energies and the structural BLA parameter is shown in Figure 9 for the two lowest excitations of NEX.
The results obtained from the MD trajectories generated with the new FF show a clear correlation between BLA and excitation energies, consistently with the previous findings in the literature.[34,37] In particular, for the lowest excitation (S1), deformations of the conjugated structure account for more than 80% of the total spread, with a coefficient of correlation
R50.802. The same analysis performed on trajectories obtained with GAFF, instead, shows a much less clear correla-tion, which implies that in these simulations other structural distortions, different from the elongation/contraction of the conjugated chain, are responsible for the largest changes in the excitation energies of the system.
As a further analysis, in Supporting Information Table S5, we summarize the calculated oscillator strengths of the first three transitions as obtained from the average of the same sets of 1000 geometries for each carotenoid, together with the values obtained on the DFT optimized structures.
The average values are in very good agreement with those obtained on the DFT geometries, showing that the geometri-cal fluctuations determined by the new FF do not change the nature of the three excitations, in particular preserving the dark and bright characters of the S1 and S2 excitations, respectively.
Figure 7. TDA TPSS/6-311G(d) transition energies (eV) of the lowest three excited states of the four carotenoids considered in this study obtained using the geometries from the MD@New_FF. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The trends obtained for the oscillator strengths are mirrored in the results on the dipole strengths (squared module of the electric transition dipole moment) as shown in Figure 10 for the three excited states of NEX (Supporting Information Table S6). Also in this case, the results obtained from the simulation performed with the new FF are very consistent with those obtained on the DFT geometries as opposed to those obtained from the GAFF dynamics. In particular, GAFF results show a significant decrease in the magnitude of the S2 dipole strength, with a corresponding increase in magnitude for the, supposedly dark, S1 state; a much clear separation between the two states is, instead, obtained with the new FF.
From all these results, it is possible to characterize the effects of temperature and structural motions on the elec-tronic excitations of carotenoids.[52] It is clear that the aver-aged excitation energies of the three lowest states are all red-shifted with respect to the values computed on the optimized structures, whereas their nature (in terms of transition dipole strengths) does not significantly change. This generalized red-shift is correlated to the “elongated” structures of the systems as commented in Structural properties section. More in detail, the first and third states seem to depend more strongly on the geometrical distortions occurring during the MD simula-tions, presenting broader distributions in excitation energies with respect to the second excitation. The much narrower dis-tribution of S2 has to be related with its composition domi-nated by the HOMO–LUMO transition.
Summary
One of the most common strategies used to describe the effects of environmental and structural fluctuations on elec-tronic processes in complex systems is to combine classical MD simulations with QM calculations of the electronic process of interest. The accuracy of this strategy, however, is largely affected by the FF used to describe fluctuations in both
intra-Figure 9. Correlation between the two lowest excitation energies and the BLA of NEX. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
and intermolecular degrees of freedom. The intramolecular aspect is particularly important when one is interested in sim-ulating electronic excitations which are largely coupled to structural deformations. Here, we have shown that an accurate description of this coupling can indeed be obtained still keep-ing a classical description of the temperature-dependent fluc-tuations by using properly parameterized MM FF. Here, the focus has been placed on the four carotenoids found in LH complexes of plants, namely NEX, VIO, LUT, and ZEA. For all of them, a newly developed FF has been proposed and the resulting MD simulations have been compared with the paral-lel ones based on a standard GAFF. The obtained results show that the most important structural properties, like backbone CC bond lengths and BLA, described by the new FF are in very good agreement with the reference DFT values, whereas the corresponding picture obtained with GAFF shows clear limits owing to an overlocalization into two separated single and double bonds. This increased accuracy in the description of the structural fluctuations is clearly reflected in the analysis of the three lowest singlet excitations. Both the expected energy ordering and the different dark (S1 and S3) and bright (S2) natures are kept during the MD based on the new FF, whereas a strong unphysical mixing is found when GAFF structures are used. This study represents the first complete determination of MM FF specifically optimized to describe the structural effects on the excitation properties of the carotenoids present in LH PPC. Its application to both energy transfer and photoprotec-tion processes should allow to achieve a detailed analysis of the important effects that the temperature-dependent fluctua-tions are known to have on both the efficiency and the robustness of these fundamental processes.
Keywords: force field
carotenoidselectronic excitation light-harvestingHow to cite this article: I. G. Prandi, L. Viani, O. Andreussi, B. MennucciJ. Comput. Chem.2016, DOI: 10.1002/jcc.24286
]
Additional Supporting Information may be found in the online version of this article.[1] T. Polivka, H. Frank,Acc. Chem. Res.2010,43, 1125.
[2] H. A. Frank, R. J. Cogdell,Photochem. Photobiol.1996,63, 257. [3] H. A Frank, G. W. Brudvig,Biochemistry2004,43, 8607.
[4] N. E. Holt, D. Zigmantas, L. Valkunas, X.-P. Li, K. K. Niyogi, G. R. Fleming,Science2005,307, 433.
[5] R. Croce, H. van Amerongen, J. Photochem. Photobiol. B 2011, 104, 142.
[6] L. Dall’Osto, C. Lico, J. Alric, G. Giuliano, M. Havaux, R. Bassi,BMC Plant Biol.2006,6, 32.
[7] F. G. Xiao, L. Shen, H.-F. Ji,Biochem. Biophys. Res. Commun.2011,414, 1.
[8] E. Formaggio, G. Cinque, R. Bassi,J. Mol. Biol.2001,314, 1157. [9] B. J. Pogson, K. K. Niyogi, O. Bjorkman, D. DellaPenna,Proc. Natl. Acad.
Sci. USA1998,95, 13324.
[10] H. Lokstein, L. Tian, J. E. W. Polle, D. DellaPenna,Biochim. Biophys. Acta Bioenerg.2002,1553, 309.
[11] Z. Liu, H. Yan, K. Wang, T. Kuang, J. Zhang, L. Gui, X. An, W. Chang,
Nature2004,428, 287.
[12] J. Standfuss, A. C. Terwisscha van Scheltinga, M. Lamborghini, W. Kuhlbrandt,€ EMBO J.2005,24, 919.
[13] Y. Umena, K. Kawakami, J.-R. Shen, N. Kamiya, Nature 2011,
473, 55.
[14] X. Pan, Z. Liu, M. Li, W. Chang,Curr. Opin. Struct. Biol.2013,23, 515. [15] A. Damjanovic´, I. Kosztin, U. Kleinekath€ofer, K. Schulten,Phys. Rev. E,
2002,65, 031919.
[16] C. Curutchet, J. Kongsted, A. Mu~noz-Losa, H. Hossein-Nejad, G. D. Scholes, B. Mennucci,J. Am. Chem. Soc.2011,133, 3078.
[17] S. Shim, P. Rebentrost, S. Valleau, A. Aspuru-Guzik,Biophys. J. 2012,
102, 649.
[18] M. Aghtar, J. Str€umpfer, C. Olbrich, K. Schulten, U. Kleinekathofer,€ J. Phys. Chem. B2013,117, 7157.
[19] S. Jurinovich, C. Curutchet, B. Mennucci, Chemphyschem 2014, 15, 3194.
[20] K. Ogata, T. Yuki, M. Hatakeyama, W. Uchida, S. Nakamura, J. Am. Chem. Soc.2013,135, 15670.
[21] C. P. van der Vegte, J. D. Prajapati, U. Kleinekathofer, J. Knoester, T. L.€ C. Jansen,J. Phys. Chem. B2015,119, 1302.
[22] S. Jurinovich, L. Viani, I. G. Prandi, B. Mennucci, Phys. Chem. Chem. Phys.2015,17, 14405.
[23] L. Viani, C. Curutchet, B. Mennucci,J. Phys. Chem. Lett.2013,4, 372. [24] C. Olbrich, J. Str€umpfer, K. Schulten, U. Kleinekathofer,€ J. Phys. Chem.
Lett.2011,2, 1771.
[25] L. Viani, M. Corbella, C. Curutchet, E. J. O’Reilly, A. Olaya-Castro, B. Mennucci,Phys. Chem. Chem. Phys.2014,16, 16302.
[26] J. Wang, R. M. Wolf, J. W. Caldwell, P. Kollman, D. Case, J. Comput. Chem.2004,25, 1157.
[27] J. Wang, W. Wang, P. Kollman, D. Case,J. Mol. Graph. Model.2006,25, 247.
[28] D. Bovi, A. Mezzetti, R. Vuilleumier, M. P. Gaigeot, B. Chazallon, R. Spezia, L. Guidoni, Phys. Chem. Chem. Phys.2011, 13, 20954.
[29] M. Macernis, J. Sulskus, C. D. P. Duffy, A. V. Ruban, L. Valkunas,J. Phys. Chem. A2012,116, 9843.
[30] M. Macernis, J. Sulskus, S. Malickaja, B. Robert, L. Valkunas, J. Phys. Chem. A2014,118, 1817.
[31] M. M. Mendes-Pinto, E. Sansiaume, H. Hashimoto, A. Pascal, A. Gall, B. Robert,J. Phys. Chem. B2013,117, 11015.
[32] W. F. Beck, M. M. Bishop, J. D. Roscioli, S. Ghosh, H. Frank,Arch. Bio-chem. Biophys.2015,572, 175.
[33] O. Andreussi, S. Knecht, C. M. Marian, J. Kongsted, B. Mennucci, J. Chem. Theory Comput.2015,11, 655.
[34] S. Knecht, C. M. Marian, J. Kongsted, B. Mennucci,J. Phys. Chem. B 2013,117, 13808.
[35] C. M. Marian, N. Gilka,J. Chem. Theory Comput.2008,4, 1501. [36] J. P. Gotze, W. Thiel,€ Chem. Phys.2013,415, 247.
[37] E. Coccia, D. Varsano, L. Guidoni,J. Chem. Theory Comput.2014, 10, 501.
[38] L. Zhang, D. A. Silva, Y. Yan, X. Huang,J. Comput. Chem. 2012, 33, 1969.
[39] M. Ceccarelli, P. Procacci, M. Marchi,J. Comput. Chem.2003,24, 129. [40] J. Cerezo, J. Zu~niga, A. Bastida, A. Requena, J. P. Ceron-Carrasco,Phys.
Chem. Chem. Phys.2013,15, 6527.
[41] D. A. Case, T. E. Cheatham, T. A. Darden, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Goetz, I. Kolossvary, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, V. B. C. Sagui, T. Luchko, S. Gusarov, A. Kovalenko, P. A. Kollman, Amber12, University of Califor-nia, San Francisco,2012.
V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels,O. Farkas, J. B. Foresman, J. V.€ Ortiz, J. Cioslowski, D. J. Fox, Gaussian09, Revision A.1; Gaussian, Inc., Wallingford, CT,2009.
[43] C. I. Bayly, P. Cieplak, W. D. Cornell, P. A. Kollman,J. Phys. Chem.1993,
97, 10269.
[44] J. Tao, J. Perdew, V. Staroverov, G. Scuseria,J. Chem. Phys.2003,91, 146401.
[45] D. A. Case, T. A. Darden, T. E. Cheatham, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Gotz, I. Kolossv€ ary, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G.Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko, P. A. Kollman, in AmberTools13 Reference Manual, San Francisco,2012.
[46] C. G. Mayne, J. Saam, K. Schulten, E. Tajkhorshid, J. C. Gumbart, J. Comput. Chem.2013,34, 2757.
[47] J. M. Seminario,Int. J. Quantum Chem.1996,60, 1271. [48] W. Humphrey, A. Dalke, K. Schulten,J. Mol. Graph.1996,14, 33. [49] Minnesota Database of Frequency Scale Factors for Electronic Model
Chemistries, Version 3 Beta 2 (accessed October 15,2014). http://t1. chem.umn.edu/freqscale/index.html
[50] I. M. Alecu, J. Zheng, Y. Zhao, D. G. Truhlar,J. Chem. Theory Comput. 2010,6, 2872.
[51] J. H. Starcke, M. Wormit, J. Schirmer, A. Dreuw,Chem. Phys.2006,329, 39.
[52] R. Spezia, C. Zazza, A. Palma, A. Amedei, M. Aschi,J. Phys. Chem. A 2004,108, 6763.


![Figure 9. Correlation between the two lowest excitation energies and the BLA of NEX. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]](https://thumb-us.123doks.com/thumbv2/123dok_us/8195803.2172703/9.918.114.819.92.406/figure-correlation-lowest-excitation-energies-color-available-wileyonlinelibrary.webp)
