LEABHARLANN CHOLAISTE NA TRIONOIDE, BAILE ATHA CLIATH TRINITY COLLEGE LIBRARY DUBLIN OUscoil Atha Cliath The University of Dublin
Terms and Conditions of Use of Digitised Theses from Trinity College Library Dublin
Copyright statement
All material supplied by Trinity College Library is protected by copyright (under the Copyright and Related Rights Act, 2000 as amended) and other relevant Intellectual Property Rights. By accessing and using a Digitised Thesis from Trinity College Library you acknowledge that all Intellectual Property Rights in any Works supplied are the sole and exclusive property of the copyright and/or other I PR holder. Specific copyright holders may not be explicitly identified. Use of materials from other sources within a thesis should not be construed as a claim over them.
A non-exclusive, non-transferable licence is hereby granted to those using or reproducing, in whole or in part, the material for valid purposes, providing the copyright owners are acknowledged using the normal conventions. Where specific permission to use material is required, this is identified and such permission must be sought from the copyright holder or agency cited.
Liability statement
By using a Digitised Thesis, I accept that Trinity College Dublin bears no legal responsibility for the accuracy, legality or comprehensiveness of materials contained within the thesis, and that Trinity College Dublin accepts no liability for indirect, consequential, or incidental, damages or losses arising from use of the thesis for whatever reason. Information located in a thesis may be subject to specific use constraints, details of which may not be explicitly described. It is the responsibility of potential and actual users to be aware of such constraints and to abide by them. By making use of material from a digitised thesis, you accept these copyright and disclaimer provisions. Where it is brought to the attention of Trinity College Library that there may be a breach of copyright or other restraint, it is the policy to withdraw or take down access to a thesis while the issue is being resolved.
Access Agreement
By using a Digitised Thesis from Trinity College Library you are bound by the following Terms & Conditions. Please read them carefully.
Electronic and Lattice Structure of the
Strongly Correlated Transition Metal
Oxides Fe3 0 4
and LaSr2
Mn2 0 7
: a hybrid
density functional study
Andrew Daniel Rowan
A Thesis submitted to
The University o f Dublin
Trinity College
for the degree of
Doctor o f Philosophy
SCHOOL OF PHYSICS
TRINITY COLLEGE DUBLIN
rJ^T R IN IT Y C O L L E G E ^
0 5 AUG 20 0 7
D eclaration
This thesis is su b m itted to th e U niversity of D ublin by th e undersigned for th e degree of D octor of Philosophy. T his thesis has no t been sub m itted as an exercise for a degree a t th is or any o th er university. T h e work carried ou t w ithin is entirely th e c a n d id a te ’s own. T he L ibrary of th e U niversity of D ublin, T rinity College, m ay lend or copy this thesis up o n recjuest.
S um m ary
T h is th esis is a th e o re tic a l s tu d y o f th e e le ctro n ic a n d la ttic e s tr u c tu r e of th e inverse sp in el m a g n e tite (Fe3 0 4) a n d th e h alf-d o p e d b ilay ered m a n g an - ite sy ste m L aS r2M n2 0 7. B o th sy ste m s are s tro n g ly c o rre la te d tra n s itio n m e ta l oxides e x h ib itin g a rich v a rie ty o f p h y sical b e h a v io u rs, d u e to th e co m p lex in te ra c tio n s o f ch arg e, sp in , o rb ita l a n d la ttic e d egrees of freedom . A h y b rid d e n sity fu n c tio n a l a p p ro a c h is a d o p te d in o rd e r to c ircu m v en t th e w ell-know n difficulties of tr a d itio n a l d e n sity fu n c tio n th e o ry a n d H a rtre e - Fock th e o ry in a c c u ra te ly d e sc rib in g th e electro n ic s tr u c tu r e o f such stro n g ly c o rre la te d tra n s itio n m e ta l oxides.
s tro n g e le c tro n re p u lsio n a n d s tro n g e le c tro n co upling to th e la ttic e , lead in g to p o la ro n ic b e h a v io u r, a n d t h a t b o th fa c to rs m u st b e a d e q u a te ly a c c o u n te d for in an y th e o re tic a l c o n sid e ra tio n of m a g n e tite physics.
T h e th e sis th e n in v e stig a te s th e h alf-d o p ed b ilayer m a n g a n ite L a S r2A'In2 0 7, w hich is of in te re s t d u e to its colossal m a g n e to re sista n c e a n d s tr u c tu r a l sim ila rity to th e h ig h -te m p e ra tu re s u p e rc o n d u c tin g c u p ra te s. T h e e x a c t a n ti fe rro m a g n e tic g ro u n d s ta te is e stab lish ed , a n d evidence is fo u n d of e x tre m e tw o -d im e n sio n a l c h a ra c te ristic s, in clu d in g a su p p ressed b u t fin ite d e n sity of s ta te s a t th e F erm i energy. T h is fe a tu re re m a in s r o b u s t even for cal c u la tio n s w ith u p to 60% H a rtree-F o ck ex ch an g e m ixing, a n d for a b -in itio o p tim is a tio n o f th e la ttic e geom etry. T h is is c o n siste n t w ith p h o to e m issio n e x p e rim e n ts w hich p o in t to w a rd s th e ex iste n c e of a ‘p se u d o g a p ’ p h ase in
A ck n ow led gem en ts
F irst and forem ost, I would like to warm ly th a n k my supervisor Dr. C harles P a tte rso n for all th e help and su p p o rt I ’ve received during th e course of th is work. T h e door to C harles’ office has always been open for all my questions and general ram blings, and all his friendly and p atien t advice and suggestions are deeply appreciated. I am p artic u la rly grateful to C harles for his u n d erstan d in g and encouragem ent regarding th e M asters degree which I sim ultaneously underto o k during the course of th is thesis, and his su p p o rt in dealing w ith th e Powers T h a t Be to enable th is crazy plan of mine to becom e a reality - it was well w orth the effort! Similarly, I would also like to acknowledge the super folks in th e D epartm ent of M athem atical Physics a t NUI Alaynooth for all th eir help and encouragem ent during th a t leg of my g ra d u a te stu d en t journey - in p articu lar Professor D anny Heffernan and Professor C harles Nash.
I would also like to acknowledge the hospitality of Professor R od B a rtle tt and th e Q u an tu m T heory P ro ject during my very enjoyable stay a t the U niversity of F lorida in sunny Gainesville, w here some of this work was carried out. T h an k s nm st go to C harles again for m aking this happen, and to th e Ti'inity T ru st for a travel award.
I would like to th a n k th e denizens of Office 2.21/3 for all th e coffee and stim u latin g conversations th a t m ade for such a pleasant working envi ronm ent, and for th eir benevolent tolerance of my occasional musings on a b stra c t topological nonsense. T h e sam e goes to th e Luce Hall footballers for m any an enjoyable kick-about which always brightened up a Friday.
To all my wonderful friends in D ublin for all th e good tim es and m oral su p p o rt - i t ’s been a privilege and a top laugh to hang ou t w ith you during my g ra d u a te stu d e n t years.
C o n ten ts
1 I n tr o d u c tio n 1
2 Q u a n tu m M a n y -B o d y T h e o r y 5
2.1 In tro d u c tio n ...
5
2.2 Hartree-Fock T h e o r y ...
8
2.3 Density Functional Theory ...
10
2.4 Structure O p tim isatio n ...
19
3 M a g n e t it e 23
3.1 In tro d u c tio n ... 23
3.2 Basic Electronic S tr u c tu r e ...
26
3.3 Early E x p e rim en t... 26
3.4 Recent E x p e rim e n t... 29
3.5 Low Tem perature Phase Results ... 35
3.5.1 Structure Optimisation ... 51
3.6 Higli Tem perature Phase R e s u l t s ... 55
3.6.1
F d3m
3 0 % ... 56
3.6.2 2 0 % ... 62
3.6.3 30% Structure O p tim isatio n ... 65
3.6.4 20% Structure O p tim isatio n ... 68
4 M a n g a n ites
82
4.1 In tro d u c tio n ...
82
4.1.1 B a c k g ro u n d ...
82
4.1.2 High-Temperature Superconducting C u p r a te s ...
86
4.1.3 Colossal M agnetoresistive M a n g a n ite s ... 89
4.2 R e su lts... 102
List o f Figures
2.1 D ia g ra m m a tic re p re s e n ta tio n o f th e in te ra c tin g -p a rtic le p ro p a g a to r G (d o u b le line) in te rm s of th e fre e -p a rtic le p ro p a g a to r Go (single line) a n d full e le c tro n self-energy E (h a tc h e d cir cle), i.e. th e series of o ne p a rtic le irre d u c ib le d ia g ra m s. Such d ia g ra m s c a n n o t b e s p lit in tw o b y rem oving a sin g le line. . . 9
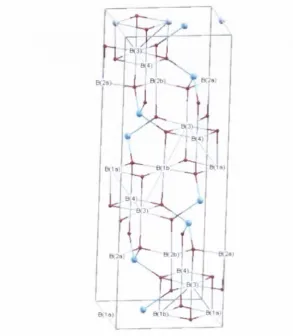
3.1 O rig in a lly p ro p o sed V erw ey ch arg e o rd e rin g schem e [8]. . . . 27 3.2 F c3 0 4 lo w -te m p e ra tu re P 2 / c cell. O c ta h e d ra l F e s ions are
la b elled as show n. T e tra h e d ra l F e^ ions are c o lo u red in cyan a n d oxygens a re in r e d ... 29 3.3 F e3 0 4 so ft x -ra y p h o to e m issio n s p e c tra . In se t: s p e c tra l edge
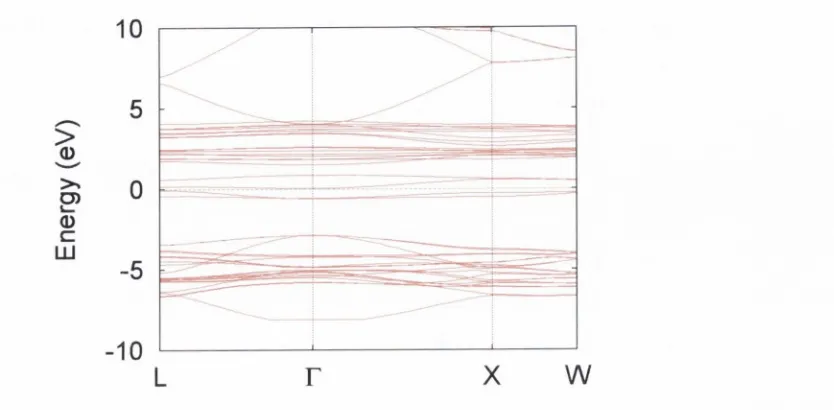
e n erg y d u rin g cooling (d o w n -trian g ies) a n d h e a tin g (u p -tria n g le s). F ro m S c h ru p p e t al [60]... 33 3.4 P 2 / c cell m a jo rity -sp in electro n ic b a n d s tr u c tu r e a t 50% ex
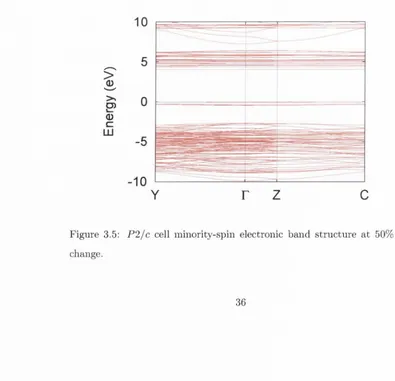
c h a n g e ... 36 3.5 P 2 / c cell m in o rity -sp in electro n ic b a n d s tr u c tu r e a t 50% ex
c h a n g e ... 36 3.6 P 2 / c cell m a jo rity -sp in electro n ic b a n d s tr u c tu r e a t 30% ex
c h a n g e ... 37 3.7 P 2 / c cell m in o rity -sp in electro n ic b a n d s tr u c tu r e a t 30% ex
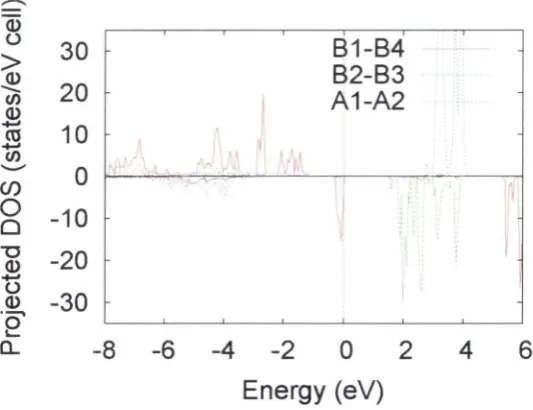
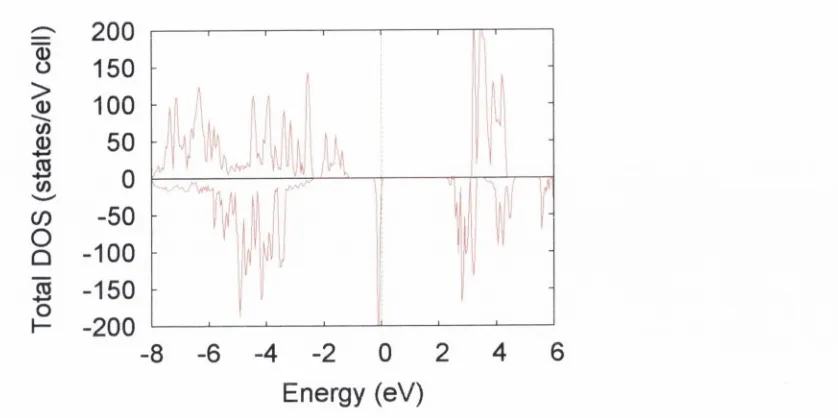
38 39 39 51 52 52 54 55 57 57 58 61 62 63 63 64 69 72 78 87 P 2 / c cell projected density of states a t 50% exchange... P 2 /c cell to tal density of sta te s a t 30% exchange... P 2 / c cell projected density of states a t 30% exchange... P 2 / c cell charge density difference plot, showing o rb ital order along a D { \ a y D { l h ) - B { \ a ) chain. C alculated a t 50% HF exchange m ixing... P 2 /c cell charge density difference plot, showing o rb ital order on Fe DA. C alculated a t 50% H F exchange m ixing... BA site orbital order, yz-plane view, a t 50% HF exchange m ixing... i? l-site orbital order on optim ised P 2 /c s tru c tu re ... Total D ensity of S tates for optim ised P 2 /c s tru c tu re ... F d 3 m cell m ajority spin b and s tru c tu re a t 30% exchange. . . F d 3 m cell m inority spin band s tru c tu re at 30% exchange. . . F d i m cell total density of sta te s a t 30% exchange... F d 3 m cell orbital order a t 30% exchange... F d 3 m cell m ajority spin band stru c tu re a t 20% exchange. . . F d 3 m cell m inority spin band stru c tu re a t 20% exchange. . . F d 3 m cell to tal density of s ta te s a t 20% exchange... F d 3 m cell pro jected density of states a t 20% exchange. O ptim ised cell to ta l density of sta te s a t 30% exchange... Total D ensity of S ta tes for optim ised s tru c tu re a t 20% exchange. F d 3 m cell w ith corresponding P I axes shown. Fe^i sites are in red, Fe^ in green and oxygen in b lu e ...
4.2 Schem atic phase diagram for cu p ra te superconductor. T he hole-doped case is on th e rig h t and the electron-doped case on th e left [19]... 87 4.3 (a) An MnOe octahedron, (b) T h e M n02 plane, identical in
stru c tu re to th e C u0 2 planes of th e h ig h -tem p eratu re super conductors. (c) T h e crystal s tru c tu re of the layered and cubic m anganite. Taken from [129] ... 92 4.4 (a) T he ion. (b) T he ion. (c,d) An iU ustration of the
concept of double exchange - th e hopping m atrix elem ent as a function of spin alignm ent, (e) T he double-exchange pre diction of th e bandw idths for th e ferrom agnetic and param agnetic cases. From Ref. [129]... 93 4.5 E xperim ental phase diagram of cubic L ai-xS r^M nO a [130]. . 94 4.6 E xperim ental phase diagram of bilayer La2_2x S ri+2xM n2 0 7
[109]. Solid points m ark m agnetic transitions determ ined from n eutron pow der-diffraction data. Open points m ark crystallographic tra n sitio n s... 94 4.7 L ai.2S ri.8M n2 0 7 A R P E S sp ectral weight versus binding en
ergy (red line). T he black line indicates the expected weight behaviour for a non-interacting theory [112]... 98 4.8 X PS sp ectra of (a) valence band (b) 0 Is (c) M n 2p of x = 0.4
and X = 0.5 taken a t 300K [139]... 100 4.9 LaSr2M n2 0 7 M n eg orb ital projected density of sta te s a t 30%
exchange (lowest energy antiferrom agnetic so lu tio n )...102 4.10 LaSr2M u2 0 7 oxygen p o rb ital p rojected density of sta te s at
30% exchange (lowest energy antiferrom agnetic solution). . . 103 4.11 L aS r2M n2 0 7 to ta l density of sta te s a t 60% exchange (lowest
4.13 O ptim ised stru c tu re projected density of sta te s a t 30% ex change... 104 4.14 L aS r2M n2 0 7 lattice. M n bilayers are shown in blue. In tra -
bilayer intercalatio n is by L a ions (purple). T h e inter-bilayer spacing contains Sr ions (grey)... 105 4.15 L aS r2M n2 0 7 electronic band stru c tu re a t 30% exchange (low
est energy antiferrom agnetic so lu tio n )... 107 4.16 L aS r2M n2 0 7 antiferrom agnetic electronic ban d stru c tu re at
60% exchange (lowest energy antiferrom agnetic solution). . . 108 4.17 L aS r2M n2 0 7 to ta l density of states a t 30% exchange (lowest
energy antiferrom agnetic solu tio n )... 108 4.18 L aSr2M n2 0 7 Fermi surface, startin g from our lowest energy
antiferrom agnetic solution a t 30% HF exchange mixing. . . . I l l 4.19 M n02 plane charge density difference a t 30% exchange. . . . 113 4.20 M n02 plane spin density difference a t 30% exchange... 113 4.21 M n 0 2 plane 3d charge density difference a t 30% exchange. . . 114 4.22 Schem atic illu stra tio n of the effects of stru c tu re optim isation
on an L aS r2M n2 0 7 bilayer... 118 4.23 C om parison of M n
Cg
states aroundEp. M n
d^2
_y2 s ta te s
for th e experim ental and optim ised stru ctu re s are shown in green and purple respectively, w ith sta te s shown in red and b lu e... 119 4.24 C om parison of O p sta te s around
Ep. Oxygen
Px, Py and
L ist o f T ables
3.1 3d-shell charge and spin populations a t 50% H F exchange. . . 40 3.2 3d-shell charge and spin populations a t 30% HF exchange. . . 40 3.3 3d-shell charge and spin populations for s tru c tu re optim ised
lo w -teniperature cell a t 50% exchange... 40 3.4 3d-shell charge and spin populations for stru c tu re optim ised
low -tem perature cell a t 30% exchange... 41 3.5 F e-0 bond lengths for th e P 2 /c and optim ised stru c tu re s w ith
m ean values for A O4 te tra h e d ra and BOq o ctah ed ra shown. . 42 3.6 Fe-Fe bond lengths for th e P 2 / c and optim ised stru ctu re s . . 43 3.7 F ractional atom ic coordinates for optim ised stru c tu re a t 50%
exchange. E xperim ental stru ctu res are taken from W right [11] and lizum i [9] ( i ta lic s ) ... 44 3.8 Fractional atom ic coordinates for optim ised stru c tu re a t 30%
exchange. E xperim ental stru ctu re s are taken from W right [11] and lizum i (italics) [ 9 ] ... 45 3.9 Fe 3d-shell charge populations from published calculations. 5
3.15 F e -0 bond lengths for th e optim ised P I stru c tu re a t 30% exchange, using our solution (Table 3.16)... 67 3.16 3c?-shell charge populations for structu re-o p tim ised cell a t 30%
exchange... 68 3.17 3d-shell charge populations for structu re-o p tim ised cell a t 30%
exchange... 69 3.18 F e -0 bond lengths for optim ised P I stru c tu re a t 20% exchange. 71 3.19 3d-shell charge populations for structu re-o p tim ised cell a t 20%
exchange... 72 3.20 3d-shell spin populations for structu re-o p tim ised cell a t 20%
exchange... 72 3.21 Irreducible representations, corresponding prim ary and sec
o ndary order p aram eters and isotropy subgroups of th e Fes0 4 F d 3 m to P I space group displacive phase tra n sitio n ... 75
4.1 C harge and spin sta te s for LaSr2M u2 0 7 a t 30% exchange. Values are shown for the experim ental and th e optim ised s tru c tu re s ...112 4.2 F ractional atom ic coordinates for experimeiital[143] and op
C h a p ter 1
In tro d u ctio n
T h e b an d th eo ry of solids has been very successful in describing th e elec tronic stru ctiu 'e of m any m aterials. C entral to th is philosophy is th e notion th a t an uneven num ber of electrons in th e u n it cell results in m etallic be- haviovu’, arising from a p artia lly filled ban d crossing the Fermi level. Very early on, however, it was pointed out by de B oer and Verwey th a t a variety of tra n sitio n m etal oxides th a t were predicted to be conductors by band th eo ry were in fact insulators [1]. M ott and Peierls ascribed this anom aly to the role of stro n g electro static repulsion in these system s, favouring electron localisation aro u n d p a rtic u la r sites. T h is electron localisation often occurs due to repulsion betw een electrons in d- and /-o rb ita ls.
given by
H = - tijcl^cja + U ^ T l i j r i i i (1.1)
<ij>,(7 i
T h e first te rm is a k in etic en erg y te rm re p re se n tin g th e h o p p in g o f ele c tro n s from s ite i to s ite j . T h e second is th e o n -site C o ulom b re p u lsio n te rm , w here riia- = is th e n u m b e r o p e ra to r.
A fu rth e r difficulty in o b ta in in g a go o d th e o re tic a l d e sc rip tio n o f m an y stro n g ly c o rre la te d tra n s itio n m e ta l oxides is th e c o m p e tin g en erg y scales p re se n t, b ey o n d sim p ly th e s tro n g M o tt-H u b b a rd in te ra c tio n U. T h e charge- tra n s fe r in te ra c tio n Upd, w hich is th e en erg y c o st for th e tra n s fe r o f a n oxygen p ele c tro n to th e n e ig h b o u rin g tr a n s itio n m e ta l ion, ca n b e sufficient to o p en a gap. Such sy ste m s are d u b b e d c h a rg e -tra n sfe r in su la to rs in th e classifica tio n schem e of Z aan en , S a w atsk y a n d A llen^. In fact, p h o to e m issio n stu d ie s p o in t to N iO g a p b ein g o i p - d c h a ra c te r [3], th u s m a k in g it a c h a rg e -tra n sfe r in su la to r in th is schem e. S tro n g e le ctro n ic co u p lin g to la ttic e d egrees of freedom c a n re s u lt in p o la ro n ic b e h a v io u r [4], a s itu a tio n w hich will b e ex am in ed fu rth e r in th is th e sis w hen we in v e stig a te th e Verwey tra n s itio n in m a g n e tite . T h e H u n d ’s ru le ex ch an g e in te ra c tio n en erg y Uex = 2 J / / , defined as th e en erg y n eed ed to flip a d e le c tro n sp in , m ay also p lay a key role in th e co n d u c tio n m ech an ism - su ch a te rm is c e n tra l in m e d ia tin g fe rro m a g n e tism in th e d o u b le ex ch an g e m o d el [5], w hich p ro v id es a first a p p ro x im a tio n to th e physics o f m a n g a n ite s like th e h a lf-d o p ed b ilay er L aS r2M u2 0 7 u n d e r s tu d y in th is work.
As is w ell-know n, th e e le c tro n ic s tr u c tu r e of m a n y s tro n g ly c o rre la te d tra n s itio n m e ta l oxides is in a d e q u a te ly d e sc rib e d by tr a d itio n a l m e a n field a b in itio a p p ro a c h e s such as H a rtre e F o c k (H F ) a n d D e n sity F u n c tio n a l T h e
o ry (D F T ), as o u tlin e d in C h a p te r 2. T h e use of h y b rid d e n sity fu n ctio n als offers th e p ro s p e c t o f a c o m p u ta tio n a lly co st effective m ean s to im prove th e re sp e c tiv e deficiencies p re se n t in H F a n d D F T , w ith o u t reco u rse to m any- b o d y p e r tu r b a tiv e ap p ro ach es, w here th e c a lc u la tio n o f th e electro n ic s tr u c tu re a n d la ttic e g e o m e try o p tim isa tio n is c o m p u ta tio n a lly p ro h ib itiv e in all b u t th e sim p le st u n it cells.
In th is th e sis, we in v e stig a te two stro n g ly c o rre la te d tra n s itio n m e ta l ox ides e x h ib itin g e x tre m e ly rich physics d u e to th e s tro n g co u p lin g o f charge, sp in , o r b ita l a n d la ttic e degrees of freedom . T h e first is th e w ell-know n inverse sp in el sy ste m m a g n e tite , Fe3 0 4, w hich u n d erg o es a m e ta l-in su la to r a n d c o n c u rre n t s y m m e try low ering s tr u c tu r a l p h a se tra n s itio n a t T ~ 125/i from a cu b ic to m onoclinic u n it cell. T h is is know n as th e Verw ey tr a n s i tio n , a n d c o n tro v e rsy h as s u rro u n d e d its e x a c t m ech an ism since its discovery over 60 y e a rs ago [6]. Fe3 0 4 re m ain s a h o t to p ic of e x p e rim e n ta l a n d th e o re tic a l re se a rc h w ith m a jo r q u estio n s o v itstanding, in clu d in g th e n a tu re of c h a r g e /o r b ita l o rd e rin g [7],[8], th e lo w -te n ip e ra tu re s tr u c tu r a l d is to rtio n [9], [10], [11] a n d th e re la tiv e roles of ele c tro n -ele c tro n c o rre la tio n [12] and e le c tro n -la ttic e co u p lin g [13] in th e Verw ey tra n s itio n m echanism . T h is w ork seeks to c la rify th e se issues using a h y b rid d e n sity fu n c tio n a l ap p ro ach .
La2_2xSri_|_2xMn207 has rem ained less explored until recent years. T his up surge of in tere st has arisen due to th e effective low -dim ensionality of these bilayer com pounds and th eir stru c tu ra l sim ilarity to the superconducting cuprates, as well as their colossal m agnetoresistive effect. We therefore fo cus on th e bilayer m anganite LaSr2M n207. T h e electronic stru c tu re and lattic e geom etry are investigated using hybrid D F T , and we find evidence of a pseudogap-like density of sta te s brought ab o u t by reduced dim ensionality in th e 2d M n02 layers which are stacked p erpendicular to th e z-axis.
C h ap ter 2
Q u a n tu m M a n y -B o d y
T h eo r y
2.1
I n t r o d u c tio n
In this work, we consider a system of N non-relativistic electrons. T he eq uation of m otion for an individual electron in th e system is th e single particle Scliroedinger ecjuation, which we w rite in tim e-dependent form.
- ( / / o + / / / ) ) V ' = 0 (2.1)
Here Hq denotes th e free p article H am iltonian and H i accounts for in ter actions, whose form we have no t yet specified. T he sta te s of th e electron
form a H ilbert space H- th e space of square integrable functions on R^. We can move to a m any-particle p icture by forming n -p article H ilbert spaces under th e o p eratio n of tensor p roduct of single p article spaces. T he collection of m ultip article sta te s for all N generates a H ilbert space called Fock space. We can define creation and annihilation operators^ and a" on th is space. T h eir action is to m ap between n-p article H ilbert spaces of different particle num bers in IF. T his allows us to define Schroedinger field
o p erato rs ■0^ = and ip — a^cf), where 4> can be a Bloch s ta te in a crystal or a localised W annier-like o rb ital in th e tight-binding lim it, of spin a and m om entum k. T hus we m ay w rite down th e two p oint correlation function
for a single p article in the A^-electron system .
i G{ x , y ) = { N m H ^ M y ) } \ N ) (2 .2)
I A^)is th e exact A^-electron ground sta te and ip^{x) is th e field o p erato r which creates an electron a t x = (r, t), while V’(y) annihilates an electron a t a tim e (y = (r',t'). T is th e tim e ordering operator:
0 ( t i ) 0 ( t 2 ) , t i > t 2
T { 0 { h ) 0 { t 2 ) ) = (2.3)
0 { t 2 ) 0 { t i ) , t 2 > t i
i G { x , y ) is in fact a G reen’s function of eciuation 2.1. T h e physical in ter p re ta tio n of th e G reen’s function is as a propagator, i.e. th e prob ab ility am p litu d e th a t, for t' > t, a.n electron added a t r a t tim e t will p ro p ag ate to r' a t tim e t' and th a t, for t > t \
a
hole added a t r' (electron rem oved) will prop ag ate to r.I n te r a c tio n s
We now consider th e exact form of interactions in th e m any-body system . T hese interactions will m odify th e m any-particle ground s ta te |A^) and th e form of th e tim e-evolution o p erato r in the in teraction picture. W^e consider a tw o-body Coulom b poten tial, w ritten in field o p erato r n o ta tio n as
V = - y d ^ r d ^ r ' i p ^ { r ' ) v c { r — (2-4)
Ferm i liquid by ad iabatically sw itching on a weak interaction. T hus th e ex citatio n s of th e Fermi gas, electrons and holes, can be continuously deform ed into so-called quasiparticles and quasiholes respectively. These quasiexcita tions will have renormalised properties owing to th e presence of interactions, such as an effective mass m* which differs from th e “b are” electron m ass m by an am ount prop o rtio n al to the stren g th of th e interaction. A physically intuitive way to think of th is is of an individual electron being screened by a p olarisation cloud as it repels neighbouring electrons. T h e incorporation of interactions introduces th e energy-dependent self energy term S (k , w) which “dresses” th e free particle p ro p a g ato r Go and contains all possible exchange- correlation interactions felt by the single electron. In an energy-m om entum representation, th e self-energy can be w ritten
E (k ,o ;) = E '( k ,w ) -f-iE " (k ,w ) (2.5)
Its real and im aginary p a rts contain all th e inform ation on energy renor m alisation and quasiparticle lifetime, respectively, of an electron w ith band energy and wavevector k. T he G reen’s function expressed in term s of the electron self-energy is given by
^
(2.0)
LJ - 6 k - S (k , w)
A related q u a n tity is the one-particle sp ectral function A (k, w) = (k, u j) + >l“ (k, w). T h e two term s in th e sum are th e one-electron addition and removal sp ectra, which can be probed directly by inverse and direct pho toem ission spectroscopy respectively. T h e one-particle spectral function is re la ted to th e G reen’s function as follows:
yl(k,w ) = - - I n i G ( k , w ) (2.7) 7T
doped M ott insulators [19] such as the bilayered colossal magnetoresistive
m anganites investigated in this thesis.
In general, the exact com putation of the self-energy E(k,u;) and the
related one-particle spectral function is a formidable task, involving sum
m ation of an infinite series which includes products of G reen’s functions
and self-energy insertions. This situation is represented diagrammatically
in Figure 2.1. A useful first order approxim ation to the full self-energy is the
Hartree-Fock self-energy. Hartree-Fock (HF) theory results from an expan
sion of the self-eziergy in term s of the bare Coulomb interaction
Vc{r —
r').
Thus higher order self-energy terms, corresponding for example to exci
tation of the electi'on hole pairs responsible for charge screening, are not
taken into account at Plartree-Fock level. The HF self-energy will be purely
real, since electrons in this single-particle theory do not feel the presence of
other
individual electrons^, resulting in an infinite lifetime for Hartree-Fock
‘quasiparticles’. In the next section, we will give a brief overview of how
Hartree-Fock theory can be implemented numerically.
We consider the non-interacting A^-electron problem. The equation of mo
tion is
Here the Hamiltonian
Hq
= {T + Vext) accounts for one-particle kinetic
energy and electron-nuclear potential energy
Vext tiut does not account for
electrons interacting with each other. The inter-electron interaction term
introduces difficulties in th a t it couples the
N
one-electron orbitals
4>i
into
an A^-electron wavefunction which is a function of 3A^ spatial coordinates
^ R a th e r th e y m ove in a n a v erag e so -called H a rtre e p o te n tia l V
2 .2
H a rtree-F o ck T h e o r y
{T+Vext)^ = E^
(2.8)Figure 2.1: Diagrammatic representation of the interacting-particle propa
gator
G
(double line) in terms of the free-partiele propagator Go (single line)
and full electron self-energy E (hatched circle), i.e. the series of one particle
irreducible diagrams. Such diagrams cannot be split in two by removing a
single line.
Ti
and
N
spin coordinates <7^. The A^-electron wavefunction in the position
basis can be obtained from the A'^-electron Fock space state vector by
Here we write
Xi
= (rj,CTj). W ithout this Coulomb interaction, the
N-electron wavefunction can be written as an antisymmetrized^ Slater deter
m inant of one-electron orbitals.
{ x i , - - - ,x n \ N ) = ,x n) (2.10)
This is normalised to one as shown:
J
dxi
■ ■ ■ J
dxNl'^ixi,
- ■
■
,X;v)|
(2.1 1)0i(ri)
4>2{ri) ■■■</>7v(ri)
0 i ( i ‘2) 02(r2) 4>N{r^2)
|^>o) =
4>i{r-s)
</>2(r3)
0iv(r3)
(2.1 2)A set of one-particle Hartree-Fock eigenvalue equations can be obtained.
Here the full Coulomb repulsion term
Veeis replaced by a mean-field Hartree
potential V// and corresponding exchange term
to account for the Pauli
exclusion principle for fermions. These terms are w ritten explicitly as
JT'i-Here n (r) is the electron density, which is given by
N
n (r) = ^ |0 i(r)p
(2.16)
i = l
This allows a self-consistent procedure to be numerically implemented; a
trial guess is made of the non-interacting electron wavefunction 'I', which
can be decomposed into the sum of
None-electron orbitals. The one-electron
Hartree-Fock equations are then solved. A new density is obtained from the
new trial wavefunction, obtained by varying the linear expansion coefficients
of the trial wavefunction. This process is repeated until the desired degree
of convergence has been obtained.
2.3
D en sity F un ction al T h eory
An alternative approach to the quantum many-body problem is afforded by
the density functional theory. The central premise is th a t the interacting
A^-electron problem with electron density n (r) can be m apped to a non
interacting problem with the same density n (r). This fictitious system of
non-interacting electrons is known as the Kohn-Sham system. Then the
(i/o
+ VW
)<^t +
j
V e x , i { r , r ’)(f)i{r’) d r ' = E(f) i{r)(2.13)
(2.14)
one-electron Kohn-Sham orbitals
(f>i
may be described by the Kohn-Sham
equations,
= ei(f)i{r)
(2.17)
with density given by equation 2.16. Here the subscript
s denotes single
electron equations.
The key shift in this viewpoint is the central point
occupied by the density n(r). This arises from the Hohenberg-Kohn The
orems [20]. The first of these dem onstrates the existence of a one-to-one
m apping between the ground state electron density and the ground state
wavefunction of a many-particle system. The second HK theorem proves
th a t the ground state density minimizes the total electronic energy of the
system.
Now the ground state functional of an interacting system of density n(r)
can be w ritten as a sum of terms
F[n] = r,[n ]
U[n] + £;,,[n]
(2.18)
Here Ts[n] is the non-interacting kinetic energy functional, while
U[n] rep
resents the Hartree-Fock energy, an exact form for which is known. The
final term is the exchange-correlation functional
Exc[n], accounting for all
other electronic interaction energy beyond Hartree-Fock level. The second
Hohenberg-Kohn theorem states th a t this functional is universal for density
n(r). However, an exact form for
Exc[n\ is in general unknown for a given
system. Thus, an approximation for the exchange-correlation energy must
be found.
The total energy for a given external potential Uexi(r) is then given by
the minimum of
i?[n] = nnn(F[n]-f-
J
d^r
V e x t i ' r )n{r))
(2-19)
This is equivalent to functional minimisation by means of the Euler-Lagrange
equation
6F
where /i =
d E / d N
is the chemical potential, and is a constant for constant
particle number
N = J
rn (r).
The Kohn-Sham potential
Vs{r) for the non-interacting system is then
u ^ (r ) = Vexti^c) + VHF{ r ) + Vxc(r) (2.2 1)
with Uxc(r) =
5 E x c [ n { r ) \ / 5 n [ r ) .Thus we know the unique Kohn-Sham
potential felt by non-interacting electrons of the same density, given some
approxim ation for
Vxc-In fact, the exchange-correlation potential
Vxcmay
be thought of roughly as a kind of D FT analogue to the electron self-energy
E, albeit local and energy-independent, unlike the full non-local, energy-
dependent electron self-energy E(k, a;).
The first and most commonly used approximation for
v^c has been the
local density approxim ation (LDA). This essentially assumes a uniform dis
tribution of electronic density. It has a relatively straight-forward extension
to spin-polarised systems by considering densities n a (r) for electrons of spin
a. This is known as the local spin density approxim ation (LSDA).
Corrections to the local density approxim ation itself are mainly classified
under the heading of gradient expansions. The idea is th at, in order to take
into account the charge inhomogeneity in the charge density of real systems,
an expansion of a functional in gradients should increase accuracy:
A^^^[n] =
J
( a ( n ( r ) )- I -6 ( n ( r ) )| V n | ^ 4-■ • •) (2.22)The most well-known such m ethod is the generalised gradient approxim ation
(GGA) for the exchange-correlation functional. Semi-empirical approaches
and fitting to small molecules have yielded popular GGA functionals such
as the Becke exchange functional [21] and the Lee-Yang-Parr correlation
functional [22].
^ L D A found to be too sm all by typically 40-50% in com parison w ith ex p erim ent [3]. T his has p artic u la r ram ifications for some strongly-correlated
“M o tt” insulators, such as th e late tran sitio n m etal monoxides, which are predicted to be m etallic w ith D F T -L D A /G G A . T hese system s usually con ta in tra n sitio n m etal or w ith p artia lly filled d shells. W hen applying a one-electron m ethod w ith an o rbital-independent p o ten tial like th e LDA, one ob tain s a p artia lly filled d band w ith m etallic-type electronic stru c tu re and itin e ra n t d electrons, often in to ta l disagreem ent w ith the experim en tally d eterm in ed M o tt insulating s ta te due to localisation of d-electrons. A sim ilar situ a tio n can arise w ith p artia lly filled /-e le c tro n shells in ra re-e arth m etal oxides.
Several a tte m p ts have been m ade to circum vent this difficulty. Self- in teraction correction (SIC) m ethods try to accovmt for th e unphysical self in teraction of the electron which is know n to occur in the LDA [23]. In exact D F T , only the highest occupied s ta te is free from self-interaction, b u t LDA has general self-interaction for all states. T his self-interaction is m ost significant for localised states; hence SIC reproduces quite well th e localised n atu re of d or / sta te s in tran sitio n m etal and ra re-e arth com pounds. How ever, SIC one-electron energies are frecjuently in strong disagreem ent w ith spectroscopic d ata.
+ U functional will be of th e general form
E L S D A + u ^ ^ a ^ ^ + E^[p^^] - E ^ c V ] (2.23)
T h e final energy functional corrects for double counting of d-orbital exchange- correlation energy. In th e absence of a specific ci-orbital basis set, equation (2.23) should reduce to th e norm al LSDA. T h e p aram eters U and J m ust be chosen for th e d-orbitals, th u s adding an em pirical elem ent to th e m ethod.
R ecent work has focussed on calculating th e H ub b ard U term from first principles using som e p e rtu rb a tiv e , frequency dependent m ethod such as the
ran d o m phase approxim ation [25], as used in th e calculation of the screened in teraction W in th e G W approxim ation. Overall, LSDA + U has been cjuite successful in reproducing experim ental results on strongly correlated tra n sitio n m etal oxides, though band stru c tu re s can be unsatisfactory, while p artia lly filled d b ands in m etallic tran sitio n m etals will also be split, re su lt ing in an unphysical in sulating s ta te for these system s.
the H u b b a rd m odel for M ott insulating system s. T h e kinetic exchange m ech
anism in th e H ubbard model gives m agnetic coupling constant J — / U ,
where t and U are th e transfer integral and on-site Coulomb interaction. U
will be large in th e unscreened HF theory, leading to a sm all J.
T h e G W M e t h o d
T h e G W m ethod [3] overcomes these problem s by considering higher-order
Feynm an diagram s which account for screening of th e bare H artree-Fock
exchange Vc, resulting in th e screened in teraction W = first considered
by H ubbard. T h is screening occurs thro u g h th e excitation of electron-hole
pairs, which are non-interacting in th e random phase approxim ation (RPA).
T h e resu ltin g electron self-energy S (k , cj) in th e G W approxim ation depends
on b o th th e frequency and wavevector of th e electron, unlike in H artree-Fock.
T his is w ritten
S (x i,ii,a ;2 ,< 2 ) = i G { x i , t i , X 2 , t 2 ) W { x i , t i , X 2 , t 2 ) (2.24)
T h e frecjuency dependence of the screened in teraction W is encoded in
th e inverse dielectric m atrix e“ ^(k,u;). T his response function m ust be first
calculated using D F T or HF eigenfunctions, and th e frequency dependence
th en m ust be tackled numerically, typically by m eans of com putationally
costly plasm on-pole or real sp ace/im ag in ary tim e approaches. Herein lies
th e considerable co m putational overhead associated w ith th e G W m ethod.
To date, th ere have been few G W studies on solids w ith stru c tu re s more
com plex th a n sim ple tran sitio n m etal m onoxides and o ther diatom ic com
pounds. T hese studies have focussed m ainly on excitation energies in the
form of quasiparticle band structures.
H y b r i d D e n s i t y F u n c ti o n a l s
An accu rate and cost effective com prom ise can be reached in th e form of
problem in LSDA by m ixing a H artree-Fock exchange energy functional
into th e to ta l LSDA energy functional. T his m akes use of the exact cancel
lation betw een Coulom b and exchange term s in H artree-Fock th eo ry and was
first proposed for sm all molecules by Becke, based on th e ad iab atic connec
tion form ula for th e exchange-correlation energy of th e K ohn-Sham density
[29]. Becke found th a t calculated ground s ta te energies for a large range of
molecules were g reatly im proved com pared to LDA alone. H ybrid function
als were subsequently found to be significantly m ore reliable th a n the best
G G A functionals for com puting atom isation enthalpies [30], geom etries and
v ib ratio n al frequencies [31]. T h e m ost com m only used hybrid functional,
sim ilar to Becke’s original scheme, is th e B3LYP functional. T his involves
th ree p aram eters A, D and C fitted to therm ocheniical d a ta - where D is
th e p ro p o rtio n of Becke’s original exchange functional [29], C is th e pro p o r
tion th e GGA correlation functional given by Lee, Yang and P a rr [22] and
A — 20% is th e percentage of H artree-Fock exchange mixing.
= (1 -
+ A E ^^ +
+ CEI:'^^
+ (1-(2.25)
T h e rem aining term s in th e above expression are th e D irac-S later
local exchange, and E ^ ^ ^ , the local density approxim ation to th e electron
gas correlation functional, following th e p aram eterisa tio n of Vosko, W ilk
and N usair [32],
Surprisingly, th is B3LYP hybrid functional is able to reproduce th e th e r
m ochem istry of tran sitio n al m etal-containing molecules, despite no tra n si
tion m etal com pounds being used in th e original d a ta [33]. D espite its
success in chem ical physics, periodic solid sta te calculations using B3LYP
have no t ap p eared until relatively recently, due m ainly to the difficulty in
tre a tin g exchange and Coulom b series to a sufficiently high level of accuracy.
Unlike LDA-I-U, which a tte m p ts to sep a rate localised d-orbitals and applies
is orbital independent in th a t it affects all Kohn-Sham orbitals in the DFT
density functional. This B3LYP hybrid approach has yielded remarkably
good band gap values in a variety of solids and insulators [34], including the
M ott insulatoi's MnO and NiO. Comparisons with comparable transitional
m etal monoxide calculations using GW [35], [36] show th a t in many instances
the band gaps obtained with B3LYP [34] have errors of similar magnitude
to GW when compared to experimental gap values. This is obtained at a
fraction of the com putational cost, and can be understood to arise from the
characteristic M ott insulator band gap errors from Hartree-Fock and DFT
cancelling each other out to some extent in B3LYP.
Thus, in GW, the bare Hartree-Fock interaction is dynamically screened
by a charge density response function, the frecjuency and wavevector de
pendent inverse dielectric m atrix e“ ^(k,a;), whereas in B3LYP, the bare
Hartree-Fock interaction is reduced in a uniform, non-frequency dependent
m anner by mixing with the D FT energy functional. To date, there have
been very few system atic direct comparisons of B3LYP and GW, or in
deed of B3LYP and LDA-f-U. A comparative study on weakly correlated
silicon [34] show good agreement between both methods and experiment.
A similar study of the prototypical strongly correlated insulator NiO in its
ferromagnetic phases show th a t the GW^ band structure based on B3LYP
wave functions is cjuite similar to the parent B3LYP band structure [37],[38].
There is a 0.2 eV broadening of the gap, from 4.1 eV to 4.3 eV, while there
are also relative shifts in the valence bands and a general upward shift in
conduction bands. This latter work also suggests th a t hybrid functionals
may be a b etter starting point for GW, since the calculation of the dielec
tric m atrix used to obtain the screened interaction
W
involves the relative
energies of valence and conduction bands obtained by a mean-field m ethod
such as D FT or HF.
func-tionals has successfully recovered th e experim ental ground s ta te m agnetic
order in a range of strongly correlated tra n sitio n m etal oxides. Studies have
been perform ed on th e high Tc p arent M o tt insulator C aC u 0 2 [39], the
colossal m agnetoresistive m anganite p aren t M o tt insulator LaM nO s [40], as
well as a range of sim ilar system s [28]. As we previously discussed, H artree-
Fock th eo ry g reatly underestim ates m agnetic coupling constants, while D F T
overestim ates. T h e above studies found th a t hybrid functionals w ith 35%
H artree-Fock m ixing, as opposed to B 3LY P’s usual 20%, in fact produced
m agnetic coupling co n stan ts in closest agreem ent w ith em pirically d eter
m ined values, while continuing to m aintain good agreem ent for ban d gaps.
In th is thesis, we ad o p t a hybrid density functional scheme sim ilar to the
above, for a range of values of H artree-Fock exchange. T his is im plem ented
in th e CRYSTAL code [41]. In order to o b tain eigenstates of th e system , it is
necessary to expand single electron wavefunctions over a set of pre-defined
basis functions. CRYSTAL uses a basis of G aussian-type orb itals of the
general form
;^GTF ^ ^ (2.26)
where dj is referred to as th e co ntraction coefficient. G aussian basis sets can
provide an ac cu rate description of electronic d istrib u tio n in b o th valence and
core states"* and are generally relatively com pact and com pu tatio n ally effi
cient. T h e quality of basis sets is a very im p o rta n t factor in determ ining
w hether self-consistent electronic stru c tu re calculations in CRYSTAL will
converge to a stab le energy m inim um . T here are a num ber of approaches
available to optim ise basis sets. We m ention the inclusion of polarisation
functions as an exam ple. T hese are functions of higher angular cjuantum
num ber th a n th e highest occupied o rb ital of th e system , which facilitate
po larisatio n effects in th e charge density. T h e inclusion of m ore basis func
tions can also aid convergence, though care m ust be taken to avoid linear
dependence errors since G aussian orbitals in general do not form a com plete
set.
2.4
S tru ctu re O p tim isation
T he d eterm in a tio n of equilibrium stru ctu re is of prim ary im portance in the
m odelling of chemical system s. Less research has been carried ou t into
periodic system s due to th e prohibitive co m p u tatio n al cost. In this section,
we will outline a modified conjugate gradient scheme, due to Schlegel [42],
which allows atom ic coordinates to be optim ised in order to locate m inim a on
th e p o ten tial energy surface. This involves com puting th e m atrix of second
derivatives of the to ta l p o ten tial energy w ith respect to atom ic positions x^.
T h e problem may be described as follows. We consider a un it cell with
N atom s. T he position of th e atom is described by th e 3-dim ensional
position vector fi. T hus we have a 3A'’-dim ensional position vector X which
describes any given atom ic arrangem ent, in som e 3A^-dimensional space S.
Every possible set of atom ic positions in the crystal can therefore be denoted
by a vector A'“ in S. T h e p o ten tial energy E is a function of 3 N variables
cycling over i atom s w ith coordinates a = 1 ,2 ,3 . T hus th ere is a
3N-dim ensional p otential energy ‘surface’ (PES), any point on which describes
the p o ten tial energy of th e lattic e for a given atom ic arrangem ent. To find an
equilibrium crystal stru c tu re , we wish to find th e p o ten tial energy m inim a
on th e PE S. T his is an exam ple of a general optim isation problem: given a
function E of ‘3 N variables Xia, we wish to find its m ininm m .
T h e conjugate g radient scheme of Schlegel [42] allows one to construct
a series of steps leading to a statio n ary p oint of th e poten tial energy E.
Here we give a brief overview of the algorithm outlined in [42]. A statio n ary
point is defined as a p oint where th e 3A^-dimensional gradient vanishes,
gi = d E / d x i — 0, yielding a gradient vector G = 0. T he first-derivative
their characterisation. Assume th a t
p
such steps have already been taken.
At step p + 1 we have the energy
E
and its gradient
at (m + 1) points
(0 < a < m < p). This information is arranged so th a t
represents the
current atomic positions,
the most recent previous point, and so on,
with
X ' ^
being the oldest. From the previous step, we also have the
n x n
m atrix of second derivatives (called the Hessian m atrix) of
E,
th a t is an
approxim ation to the true Hessian
Fij = d?E/dx^dx^.
An optim isation step consists of three parts. Firstly, the Hessian from
the previous step is corrected. Then, a search is performed for a minimum
between the current PES point and the previous point, i.e. in the direction
{X'^ —
X®). Finally, using the Hessian m atrix and minimum thus obtained,
the next estim ate of the location of the stationary point is given by
j
Here i j and
gj
are the position coordinates of the previous minimum, and
th a t of its numerically obtained gradient
g.
Thus we see th a t gradients
are evaluated every tim e the total energy is computed; the second derivative
(Hessian) m atrix is built from the gradients. At each step, a one-dimensional
minimisation using a cjuadratic polynomial is carried out, followed by a
ZN-dimensional search using the Hessian.
If a negative eigenvalue is found, its sign is reversed. This forces a steep
est descent step along the direction of the eigenvector possessing the negative
eigenvalue. At each step, the gradient vector
and the displacement vec
tor
are tested against given tolerance levels for convergence. The
root-mean-square gradient and displacement, as well as the absolute value
of the largest component of each, must fall below their respective tolerance
levels. If these four conditions are satisfied, the optim isation is considered
complete. The above algorithm is implemented in parallel in the CRYS-
P h o n o n M o d e s
The Hessian m atrix
Fij may also be used to obtain the vibrational eigen-
modes of the crystal and their associated vibrational eigenfrequencies. This
information on phonon modes can provide essential insight into physics of
the system under study. We recall th a t if there are
N
atoms in the prim
itive cell, then there are
?>N
branches to the phonon dispersion relation: 3
acoustic branches (a longitudinal LA and two transverse TA), and
“
i N
— 3
optical branches, also split into longitudinal LO and tranverse TO modes.
Thus for
P primitive cells, the crystal will have a total of ‘iN P
vibrational
degrees of freedom.
Vibrational frequencies may be calculated at the F point of the Bril-
louin zone as follows. Using the Hessian
Fij as calculated above, the mass-
weighted Hessian is calculated:
F ,
^Vai,0j{q = 0) =Ma and
M(j are the masses of atoms
a and
(3
associated with the
and
coordinates, respectively. The m atrix
is then diagonalised to ob
tain eigenvalues (phonon mode frequencies) and eigenvectors (phonon mode
atomic displacement vectors). Certain advantages accrue from calculations
performed at the
F point. TF(0) is simple to calculate from the expres
sion above, and possesses the point symm etry of the crystal. In addition,
the three acoustic modes, corresponding to translations at F, have zero fre
quency. This in fact should act as a check of the reliability of calculated
results. Results for which the three acoustic modes show non-zero frequen
cies should be discarded. Lastly, it is the F point modes which give rise to
the infrared and Ram an spectra observed in experiments.
This com putationally demanding process has been implemented in a
beta version of the CRYSTAL06 code which we have adopted in this work.
be tak en to ensure th a t th e initial crystal stru c tu re is a t a m inim um of
th e p o ten tial energy surface before v ib ratio n al calculations are carried out.
T his in effect entails a tte m p tin g phonon calculations only on cell geom etries
which have already been converged and optim ised to high accuracy, typically
w ith tolerances governing th e Coulom b and exchange integral convergence
crite ria set to 10“ ® H artree.
T h e hybrid density functionals used in this work have proven effective
for equilibrium cell geom etry optim isations [43]. T h e LDA and G G A alone
C h ap ter 3
M a g n e tite
3.1
I n tr o d u c tio n
M a g n e tite (Fe3 0 4) is th e o ld est know n m a g n e tic m a te ria l, h av in g b een d oc u m e n te d over 2500 y ears ago by th e a n c ie n t G reeks. I t has th e la rg e st n e t
m a g n e tiz a tio n o f all th e n a tu ra lly o c c u rrin g m in erals on E a rth , an d th e se
m a g n e tic p ro p e rtie s led to m a g n e tite , o r lo d esto n e, b e in g used as a n early
form of m a g n e tic com pass. T h e re m a rk a b le p h y sical p ro p e rtie s of th is m a
te ria l e n su re t h a t it co n tin u es to b e of im m en se c u rre n t in te re st. As a
h a lf-m e ta l a t ro o m te m p e ra tu re , w ith a n u n u su a lly h ig h C u rie te m p e ra tu re ,
m a g n e tite is o f technological in te re s t as a p o te n tia l so u rce o f sp in -p o la rise d
e le c tro n s in s p in tro n ic s a p p lic a tio n s [44]. M a g n e tite is also cited as a possible
n m ltife rro ic m a te ria l [45] - t h a t is, a m a te ria l w hich is sim u lta n e o u sly m a g
n e tic a n d ferro electric, re n d e rin g it of c o n sid erab le th e o re tic a l a n d p ra c tic a l
im p o rta n c e . In a d d itio n , th is sy ste m d isp lay s th e (in )fam o u s V erw ey p h a se
tra n s itio n , th e ex a c t n a tu re of w hich re m a in s co n d en sed m a tte r p h y sic s’
lo n g est r u n n in g controversy.
M a g n e tite h a s th e form al ionic fo rm u la Fe^“''[Fe^'''Fe^''']0 4. T h e re are tw o in e q u iv a le n t iron s u b la ttic e s in th e u n it cell: th e te tra h e d ra lly c o o rd in a te d
ions. T his system undergoes a phase tran sitio n a t Tjv = 860i^ from a
param ag n etic to a ferrim agnetic state. A t T y ^ 125K, th e system undergoes
an o th er phase tran sitio n , characterised by a stru c tu ra l disto rtio n and an
a b ru p t decrease in conductivity by some two orders of m agnitude. T his
class of tra n sitio n is nam ed after Eugene Verwey, who first discovered it in
Fe3 0 4 in 1939 [6]. M ore generally, F es0 4 has served as a p ro to ty p e for m any
early studies of m agnetism . Neel developed his theory of antiferrom agnetism
following studies on th is system [46], while th e nascent th eo ry of m etal-
insulator tran sitio n s in strongly correlated system s received m uch im petus
from observations of the Verwey tran sitio n , in p artic u la r the early studies
of M ott and Peierls (see [47] and references therein).
Fes0 4 belongs to th e class of spinel ferrites, as first d eterm ined by the
early x-ray studies of B ragg [48, 49]. Such system s have th e general fornm la
A D2O4, w ith a cubic cell consisting of 32 close-packed oxygen ions, witli 64
te tra h e d ra lly coordinated A -type sites and 32 o ctah e d ral Z?-type sites. 8 of
th e A -sites are occupied by Fe^+ ions while 16 of th e D-sites are occupied
by Fe^"*". O bserving th e unusually high conductivity of this m ateria l above
th e m etal-in su lato r tran sitio n , relative to sim ilar spinels such as C0 3O4 an d
M n3 0 4\ Verwey concluded th a t the correct arrangem ent of Fe ions was th e
inverse spinel stru ctu re , w hereby th e first o ctet of Fe^+ ions reside on th e
te tra h e d ra l sites, while th e second octet, along w ith th e Fe^"^ ions, reside
on th e 16 B -sites. T h is arrangem ent im m ediately suggests a sim ple conduc
tion m echanism due to hopping of th e ‘e x tra ’ Fe^"*" electron betw een closely
spaced B-sltes. T his m echanism was used by Verwey in a schem atic th e
ory of a p u ta tiv e order-disorder tran sitio n , whereby th e high te m p e ra tu re
disordered m etallic phase d em o n strated conduction by th e above channel,
w ith th e lo w -tem perature in sulating phase resulting from a localization of
conduction electrons on th e 8 B -sites. T his localization was p o stu la te d to
lead to a m ixed valence charge-ordered state, w ith a concom itant sym m etry-
lowering stru ctm 'al d isto rtio n from cubic to tetragonal. Such a scenario also
allows for th e possibility of so-called ‘orbital o rd e r’, a real space ordering of
charge carriers in certain orbitals.
Very early on, however, ciuestions were raised ab o u t the validity of th e
Verwey charge ordering model. E xperim ental studies often produced con
flicting results, in no sm all p a rt due to the poor cjuality of m agnetite sam
ples used in m any early studies, and controversy still persists to th e present
day on th e exact m echanism of the Verwey transition. In m any respects,
th is could be considered th e oldest unresolved question in condensed m a tte r
physics.
In th e next section, we will outline the basic electronic stru c tu re of this
m ateria l and discuss the influence of com peting energy scales and strong elec
tronic correlation on th e m acroscopic properties of th is system . Strongly cor
related system s such m agnetite, as well as m any o ther tran sitio n m etal ox
ides such as th e colossal m agnetoresistive m anganites and high-tem p eratu re
superco n d u ctin g cuprates, ten d to order; th is can occur in th e spin, charge
or o rb ital sectors and is a continuing hot topic of research in the physics of
condensed m a tte r. Hence m ag n etite provides us w ith a useful platform for
th e stu d y of th e general phenom enon of order in strongly correlated elec
tro n system s. We will also provide a brief overview of th e developm ent of
experim ental and theoretical research in m agnetite physics, allowing us to
identify th e historical stum bling blocks which have con trib u ted to th e con
tinued controversy over th e exact m echanism of th e Verwey tran sitio n since
its discovery over 60 years ago. T his will provide a context for our work and
it is hoped th a t our hybrid H F /D F T calculations help to shed some light on
3.2
B asic E lectron ic S tru ctu re
It is instru ctiv e to have som e heuristic p icture of tlie basic electronic stru c tu re of th is system . T h u s we consider th e configuration in term s of free Fe^"*" an d Fe^+ ions in th e ap p ro p riate crystal environm ent. In th is sim ple picture, th e tetra h e d ra l A -sites contain Fe^"*" ions, yielding a electronic configu ration. A ccording to H u n d ’s rules, th o u g h strictly valid only in th e case of a free atom , th is gives a singlet ^5 5 /2 ground s ta te term . T h e p oint group sym m etry of th e A -site is cubic; however, th ere is no crystal field splitting for the o rb ital singlet S ground sta te . T here is also no spin-orbit splitting, since th e th e o rb ital m om ent is zero. Therefore, we have a spin-only ground s ta te 5 = 5 /2 . A sim ilar situ atio n arises for th e D -site Fe^"*".
In th e case of th e o c te t of Fe^+ ions on B -sites, H u n d ’s rule filling yields a spin ground s ta te of 5 = 2. T h e n ea rest neiglibour oxygen o ctah ed ral field splits th e ground s ta te into trip ly degenerate t2g levels and an excited
sta te 6g doublet. T his allows a possible t2g o rb ital ordering to take place.
It is w orth notin g th a t th e 5 rf-electrons are arranged in a relatively high spin s ta te w hen com pared to some o th er well-known strongly correlated system s such as th e high te m p e ra tu re su perconducting cuprates, where a dP
configuration on copper yields a spin of 5 = 1/2 per Cu ion[14]. T his low spin s ta te has th e effect of m aking th e system som ew hat m ore “q u an tu m ” in n a tu re [16].
3.3
E arly E xp erim en t
T h e lite ra tu re on th is long-running su b ject is encyclopedic; in th is section, we give a b rief overview of th e relevant highlights p ertin e n t to our research.
octahedrally coordinated ions in the lo w -teinperature phase, whereby these
ions arrange in alte rn a te a/4 -sp aced (100) planes, w ith hom ovalent ions
form ing chains along th e (110) and (IlO ) directions as shown in Figure 3.1.
(t, -•
-J
•■>. ■^.010-'F igure 3.1: O riginally proposed Verwey charge ordering scheme [8].
T his charge ordering arrangem ent induces an orthorhom bic disto rtio n
w ith respect to th e high-tem p eratu re cubic cell. Such a crystal geom etry
should result in a (002) reflection visible by n eutron diffraction. T his reflec
tion was detected by H am ilton in 1958 [51] using sy n th etic single crystals
and applying a m agnetic field to pick a spin o rientation, th u s counteracting
m ultiple tw inning effects. Such tw inning effects can arise where a crystal has
a mosaic of different cubic [001] axes for spin orientations, thus exhibiting
overall isotropic behaviour. However, th e sam e p ap e r also reports experi
m ents perform ed on n a tu ra l crystals, which did no t reveal any (002) reflec
tion consistent w ith orthorhom bic sym m etry, though th e au th o r a ttrib u te d
this to th e defects present in n atu ra lly occurring m ag n etite crystals.
L ater neutron scatterin g studies by Shirane et al in 1975 [52] also failed
to detect the (002) reflection, and suggested th a t it was m erely an arte fac t
resulting from sim ultaneous reflections. T heir m agnetic intensity d istrib u
tion suggested altern ate Fe^"*" and Fe^"*" in th e a6-plane, in co n tra st to th e
[image:42.537.61.524.45.431.2]electronic and x-ray diffraction results revealed superlattice reflections incon
sistent with orthorliombic symmetry, indicating th a t the low tem perature
cell is monoclinic, in contravention of the original Verwey charge ordering
scheme.
Detailed neutron diffraction experiments on the low-tem perature struc
ture by lizumi et al [9] in 1982 proposed a monoclinic
Cc
supercell of di
mension \/2 a
X\/2 a
x2a
relative to the high-tem perature cubic cell. In
these experiments, care was taken to check for spurious multiple twinning
effects. Structural refinements were carried out using orthorhombic synmie-
try constraints on atomic positions. Besides proposing a
Cc
space group,
the authors reported no significant variation in the mean Fe-0 distances for
the Fe i?-sites, such as might accompany charge ordering. lizum i’s results
also cast doubts on a previous conjectured charge ordering model due to
Mizoguchi, which was derived on the basis of NMR measurements [53].
It seems th a t many early experimental studies of m agnetite were plagued
by the poor quality of samples employed. This stemmed in part from the
widespread use of natural crystals, which suffer from an array of imper
fections such as substitutional impurities, octahedral vacancies, dislocations
and internal stresses. Also, many synthetically produced crystals did not fol�
![Figure 3.1: Originally proposed Verwey charge ordering scheme [8].](https://thumb-us.123doks.com/thumbv2/123dok_us/1019002.616908/42.537.61.524.45.431/figure-originally-proposed-verwey-charge-ordering-scheme.webp)