metal-organic papers
m540
Sun, Zhang and Yang [Zn(C10H2O8)](NH4)28H2O DOI: 10.1107/S1600536802015994 Acta Cryst.(2002). E58, m540±m542Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
A nanoporous three-dimensional metal coordination
polymer {(NH
4)
2Zn[C
6H
2(COO)
4]
8H
2O}
nYan-Qiong Sun, Jie Zhang and Guo-Yu Yang*
State Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, People's Republic of China
Correspondence e-mail: ygy@ms.fjirsm.ac.cn
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(C±C) = 0.003 AÊ H-atom completeness 39% Disorder in solvent or counterion
Rfactor = 0.032
wRfactor = 0.097
Data-to-parameter ratio = 13.5
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2002 International Union of Crystallography Printed in Great Britain ± all rights reserved
The title compound, poly[ammonium zinc- -benzene-1,2,4,5-tetracarboxylate octahydrate] {(NH4)2Zn[C6H2(COO)4]
-8H2O}n, forms a three-dimensional network containing one-dimensional nanoporous rhomboid channels along thecaxis, accommodating guest water molecules and the ammonium cations. The dimensions of the large channel are 11.555 (2)
15.00 (1) AÊ. The network consists of [Zn(C6H2(COO)4]2ÿ
anions containing tetrahedral Zn atoms bonded to four carboxylate groups. The Zn O distances range from 1.974 (2) to 1.978 (2) AÊ. The ammonium cations and the water molecules are associated in a centrosymmetric network by intermolecular hydrogen bonds. The hydrogen bonding is essential to stabilize the crystal structure.
Comment
Aromatic polycarboxylate ligands have versatile coordination modes as they can form bridges between metallic centers, generating various molecular topologies and architectures, as well as having potential application as functional materials. They ®nd widespread application in heterogeneous catalysis, chemical absorption and magnetism. Owing to their wide-spread utility (Yaghi & Li, 1996), a considerable number of reports based on metal±aromatic polycarboxylate coordin-ation polymers have appeared over the past few years (Yaghi et al., 1997). 1,2,4,5-Benzenetetracarboxylic acid (H4btec) has
four carboxyl groups, with eight O-donor atoms that may be completely or partly deprotonated, inducing rich coordination modes and allowing novel structures with up to three-dimensional networks. It has been reported that an increase in pH results in a higher connectivity level of the ligands, which in turn leads to a higher dimensionality of the crystal structure
(Pan et al., 2001). We adjusted the pH value to 9±10 with
NH3H2O and obtained the title compound, (I).
{(NH4)2Zn[C6H2(COO)4]8H2O}n is a three-dimensional polymer containing one-dimensional nanoporous channels along thec axis, accommodating guest water molecules and the ammonium cation. All the carboxyl groups of H4btec are
deprotonated, in agreement with the IR data, in which no absorption peaks are observed around 1700 cmÿ1for COOH.
There is a strong absorption peak at 1603 cmÿ1for as(CO).
Only one type of coordination mode of btec4ÿis present in the structure. Each carboxylate group adopts a monodentate mode, coordinating one Zn atom. Therefore, each btec4ÿacts as an4-bridge, linking four Zn centers, and each Zn atom
attaches to four btec4ÿ ligands, forming a slightly distorted tetrahedron around Zn2+, with ZnÐO bond lengths ranging
from 1.974 (2) to 1.978 (2) AÊ, similar to other related ZnÐO distances (Robl, 1992). Zn2+is situated on the twofold axis and
the four coordinated O atoms are related to each other in pairs by the twofold axis; the btec4ÿphenyl ring is centrosymmetric. The closest Zn Zn distance is 5.538 (1) AÊ, indicating the lack of any direct metal±metal interaction. The macrocyclic unit [Zn4(btec)4]12ÿ, which forms a rhomboid sheet, with a
Zn Zn distance of 9.587 (1) AÊ, constitutes the basic building block of the structure. Every building block is linked together through ZnÐO bonds to generate a three-dimensional structure with a nanoporous rhomboid channel along the c axis. The dimensions of this large channel are 11.555 (2)
15.100 (3) AÊ. It accommodates ammonium cations and water molecules associated into a centrosymmetric network by intermolecular hydrogen bonds. Different kinds of hydrogen bonding are observed in the structure: (a) hydrogen bonding among water molecules [O O distance: 1.845 (2)± 3.039 (2) AÊ]; (b) hydrogen bonding of water molecules and carboxylate O atoms [O O distance: 2.725 (8)±3.028 (1) AÊ]; (c) hydrogen bonding between the ammonium cations and
carboxylate O atoms [N O distance: 2.847 (2)±3.016 (2) AÊ]; (d) hydrogen bonding between the ammonium cations and water molecules [N O distance: 2.894(2)±3.058 (2) AÊ]. The guest water molecules and ammonium cations are obviously essential to stabilize the crystal structure.
Experimental
A mixture of Zn(CH3COO)23H2O (0.043 g, 0.2 mmol),
1,2,4,5-benzenetetracarboxylic dianhydride (0.043 g, 0.2 mmol), NH3H2O
(1 ml) and H2O (6 ml) with a molar ratio of 1:1:278:1667 (solution pH
= 8±9) was heated in a stainless steel reactor with a Te¯on liner at 433 K for three days. It produced a colorless solution. After evaporation for several days at room temperature, colorless rhom-boidal crystals were obtained.
Crystal data
[Zn(C10H2O8)](NH4)28H2O Mr= 495.70
Monoclinic,C2=c a= 11.555 (2) AÊ
b= 15.300 (3) AÊ
c= 11.072 (2) AÊ
= 91.97 (3)
V= 1956.4 (7) AÊ3 Z= 4
Dx= 1.683 Mg mÿ3 Mo Kradiation Cell parameters from 50
re¯ections
= 2.2±27.5
= 1.34 mmÿ1 T= 293 (2) K Rhomboidal, colorless 0.200.190.10 mm
Data collection
Siemems SMART CCD diffractometer
'and!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin= 0.762,Tmax= 0.877
4233 measured re¯ections
2242 independent re¯ections 2015 re¯ections withI> 2(I)
Rint= 0.010 max= 27.5
h=ÿ14!14
k=ÿ19!18
l=ÿ14!14
Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.032 wR(F2) = 0.097 S= 0.98 2242 re¯ections 166 parameters
H atoms treated by a mixture of independent and constrained re®nement
w= 1/[2(F
o2) + (0.0773P)2 + 1.4324P]
whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.66 e AÊÿ3 min=ÿ0.59 e AÊÿ3
Acta Cryst.(2002). E58, m540±m542 Sun, Zhang and Yang [Zn(C10H2O8)](NH4)28H2O
m541
metal-organic papers
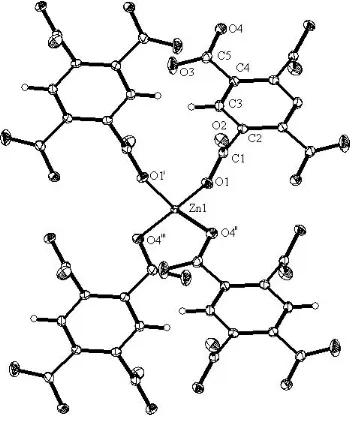
Figure 1
The Zn2+coordination environment in the title compound and the
atom-labeling scheme, Displacement ellipsoids are shown at the 30% probability level [symmetry codes: (i) 1ÿx, y,ÿz+1
2; (ii)x+12,yÿ12,
z; (iii)ÿx+1
2,yÿ12,ÿz+12].
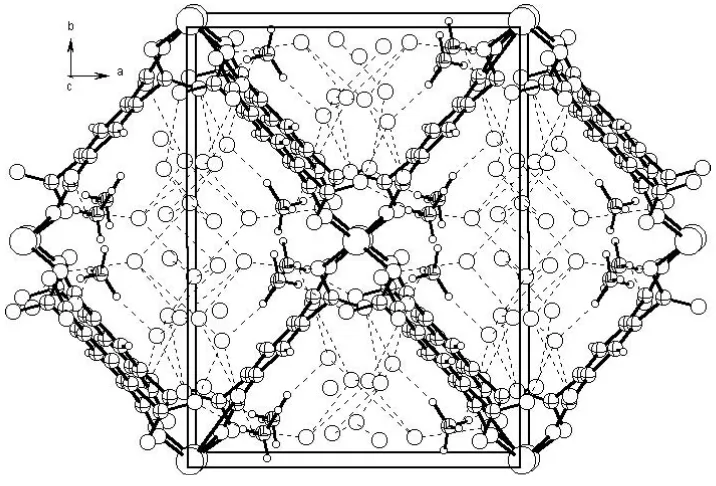
Figure 2
metal-organic papers
m542
Sun, Zhang and Yang [Zn(C10H2O8)](NH4)28H2O Acta Cryst.(2002). E58, m540±m542Table 1
Selected geometric parameters (AÊ,).
Zn1ÐO4i 1.9744 (14)
Zn1ÐO1 1.9789 (14)
O1ÐC1 1.276 (2)
O2ÐC1 1.235 (3)
O3ÐC5 1.237 (3)
O4ÐC5 1.268 (2)
O4iiÐZn1ÐO4i 123.02 (9)
O4iiÐZn1ÐO1 103.79 (6)
O4iÐZn1ÐO1 104.91 (6)
O1iiiÐZn1ÐO1 117.40 (8)
O2ÐC1ÐO1 124.74 (17)
O3ÐC5ÐO4 123.50 (18)
Symmetry codes: (i)1
2ÿx;yÿ12;12ÿz; (ii)12x;yÿ12;z; (iii) 1ÿx;y;12ÿz.
The phenyl ring and ammonium H atoms were located in a difference Fourier map and their positions and isotropic displacement parameters were re®ned. Atom O3wis equally disordered over two positions (O3wÐO3w0 distance: 1.523 AÊ). The occupancy of atom
O5wre®ned to 0.496 and was ®xed at 0.5. The re®nement is unstable if the occupancy of O5wis set to 1. The H atoms of the water O atoms were not located in the difference Fourier map, and were not included, owing to the complexities of the structure.
Data collection:SMART(Bruker, 1999); data reduction:SAINT
(Bruker, 1999);cell re®nement:SMART; data reduction:SHELXTL
(Bruker, 1997); program(s) used to solve structure: SHELXS97
(Sheldrick, 1990); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); molecular graphics:SHELXTL; software used to prepare material for publication:SHELXTL.
This work was supported by the Ministry of Finance of China, the National Science Foundation of China (Grant No.20171045) and the Talents Program of the Chinese Academy of Sciences.
References
Bruker. (1997). SHELXTL. Version 5.10. Bruker AXS Inc., Madison, Wisconsin, USA.
Bruker. (1999).SMART(Version 5.054) andSAINT(Version 5.054). Bruker AXS Inc., Madison, Wisconsin, USA.
Pan, L., Frydel, T., Sander, M. B., Huang, X. Y. & Li, J. (2001).Inorg. Chem.40, 1271±1283.
Robl, C. (1992).Mater. Res. Bull.27, 99±107. Sheldrick, G. M. (1990).Acta Cryst.A46, 467±473.
Sheldrick, G. M. (1996).SADABS. University of GoÈttingen, Germany. Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany. Yaghi, O. M., Li, H. L., Groy & T. L. (1996).J. Am. Chem. Soc.118, 9096±9101. Yaghi, O. M., Charles, E. D., Li, G. M., Li & H. L. (1997).J. Am. Chem. Soc.
supporting information
sup-1
Acta Cryst. (2002). E58, m540–m542
supporting information
Acta Cryst. (2002). E58, m540–m542 [doi:10.1107/S1600536802015994]
A nanoporous three-dimensional metal coordination polymer
{(NH
4)
2Zn[C
6H
2(COO)
4]
·
8H
2O}
nYan-Qiong Sun, Jie Zhang and Guo-Yu Yang
S1. Comment
Aromatic poly-carboxylic ligands have a versatile coordination modes as they can form bridges between metallic centers,
generating various molecular topologies and architectures, as well as having potential application as functional materials.
They find widespread application in heterogeneous catalysis, chemical absorption and magnetism. Owing to their
widespread utility (Yaghi et al., 1996), a considerable number of reports based on metal–aromatic poly-carboxylic
coordination polymers have appeared over the past few years (Yaghi et al., 1997). 1,2,4,5-Benzenetetracarboxylic acid
(H4btec) has four carboxyl groups, eight O-donor atoms that may be completely or partly deprotonated, inducing rich
coordination modes and allowing novel structure with higher dimensions. It has been reported that an increase in pH
results in a higher connectivity level of the ligands, which in turn leads to a higher dimensionality of the crystal structure
(Pan et al., 2001). We adjusted the PH value to 9 ~10 by NH3·H2O and fortunately obtained the title compound, (I).
{(NH4)2Zn[C6H2(COO)4]·8H2O}n is a three-dimensional polymer containing one-dimensional nanoporous channels
along the c axis with guest water molecules and the ammonium cation. All the carboxyl groups of H4btec are
deprotonated, in agreement with the IR data in which no absorption peaks are observed around 1700 cm−1 for COOH.
There is a strong absorption peak at 1603 cm−1 for υ as(CO).
Only one type of coordination mode of btec4− is present in the structure. Each carboxylate group adopts a monodentate
chelating mode, coordinating one Zn atom. Therefore, each btec4− acts as a µ4-bridge linking four Zn centers and each
central Zn atom attaches to four btec4− ligands, forming a slightly distorted tetrahedron around Zn2+, with Zn···O bond
lengths ranging from 1.974 (2) to 1.978 (2) Å, similar to other related Zn···O distances (Robl.,1992). Zn2+ is situated on
the twofold axis and the four O atoms in a pair are related to each other by the twofold axis; the btec4− phenyl ring is
centrosymmetric. The closest Zn···Zn distance is 5.538 (1) Å, indicating the lack of any direct metal–metal interaction.
The macrocyclic unit [Zn4(btec)4]12−, which forms a rhombus sheet, with a Zn···Zn distance of 9.587 (1) Å, constitutes the
basic building block of the structure. Every building block is linked together through Zn···O bonds to generate a
three-dimensional structure with a nanoporous rhomboid channel along the c axis. The dimensions of this large channel are
11.555 (2) Å × 15.100 (3) Å. It accommodates ammonium cations and water molecules associated into a
centrosymmetric network by intermolecular hydrogen bonds. Different kinds of hydrogen bonding are observed in the
structure: (a) hydrogen bonding among water molecules [O···O distance: 1.845 (2)–3.039 (2) Å]; (b) hydrogen bonding
of water molecules and carboxylate oxygen atoms [O···O distance: 2.725 (8)–3.028 (1) Å]; (c) hydrogen bonding between
the ammonium cation and carboxylate oxygen atoms [N···O distance: 2.847 (2)–3.016 (2) Å]; (d) hydrogen bonding
between the ammonium cation and water molecules [N···O distance:2.894 (2)–3.058 (2) Å]. The guest water molecules
supporting information
sup-2
Acta Cryst. (2002). E58, m540–m542
S2. Experimental
A mixture of Zn(CH3COO)2·3H2O (0.043 g, 0.2 mmol), 1,2,4,5-benzenetetracarboxylic dianhydride (0.043 g, 0.2 mmol),
NH3·H2O (1 ml) and H2O (6 ml) with a molar ratio of 1:1:278:1667 (solution PH=8 ~9) was heated in a stainless steel
reactor with a Teflon liner at 433 K for three d. It produced a colorless solution. After evaporation for several days at
room temperature, colorless diamond-like crystals were obtained.
S3. Refinement
The phenyl ring and ammonium hydrogen atoms were located in a difference Fourier map and their positions and
isotropic displacement parameters were refined. Atom O3w is equally disordered over two positions (O3w—O3w′
distance: 1.523 Å). The occupancy of atom O5w is 0.496 and assigned to 0.5. The refinement is unstable if the occupancy
of O5w is assigned to 1. The hydrogen atoms of the water O atoms were not located in the difference Fourier map, and
supporting information
sup-3
[image:6.610.130.483.71.492.2]Acta Cryst. (2002). E58, m540–m542
Figure 1
The Zn2+ coordination environment in the title compound and the atom-labeling scheme, Displacement ellipsoids are
shown at the 30% probability level [symmetry codes: (i) 1 − x, y, −z + 0.5; (ii) x + 0.5, y − 0.5, z; (iii) −x + 0.5, y − 0.5, −z
supporting information
sup-4
[image:7.610.126.484.70.313.2]Acta Cryst. (2002). E58, m540–m542
Figure 2
Packing diagram, viewed down the [001] diagonal of the unit cell, showing hydrogen-bond interactions.
poly[Ammonium Zinc-µ-benzene-1,2,4,5- tetracarboxylato-octahydrate
Crystal data
[Zn(C10H2O8)](NH4)2·8H2O
Mr = 495.70 Monoclinic, C2/c Hall symbol: -C 2yc a = 11.555 (2) Å b = 15.300 (3) Å c = 11.072 (2) Å β = 91.97 (3)° V = 1956.4 (7) Å3
Z = 4
F(000) = 1032 Dx = 1.683 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 50 reflections θ = 2.2–27.5°
µ = 1.34 mm−1
T = 293 K
Diamond, colorless 0.20 × 0.19 × 0.10 mm
Data collection
Siemems SMART CCD diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996) Tmin = 0.762, Tmax = 0.877
4233 measured reflections 2242 independent reflections 2015 reflections with I > 2σ(I) Rint = 0.010
θmax = 27.5°, θmin = 2.2°
h = −14→14 k = −19→18 l = −14→14
Refinement Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.032
wR(F2) = 0.097
S = 0.98
2242 reflections 166 parameters 0 restraints
supporting information
sup-5
Acta Cryst. (2002). E58, m540–m542
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0773P)2 + 1.4324P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.66 e Å−3
Δρmin = −0.59 e Å−3
Special details
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of
F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R
-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be
even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq Occ. (<1)
Zn1 0.5000 0.494950 (16) 0.2500 0.01785 (13)
O1 0.38370 (12) 0.56214 (9) 0.15306 (13) 0.0270 (3)
O2 0.52526 (13) 0.65120 (11) 0.09969 (16) 0.0366 (4)
O3 0.1452 (2) 0.84669 (13) 0.27795 (15) 0.0538 (5)
O4 0.08978 (13) 0.93339 (9) 0.12755 (14) 0.0292 (3)
C1 0.42225 (17) 0.62977 (12) 0.10034 (17) 0.0235 (4)
C2 0.33176 (16) 0.68962 (11) 0.04435 (17) 0.0221 (4)
C3 0.27625 (18) 0.74539 (13) 0.12211 (18) 0.0257 (4)
C4 0.19476 (16) 0.80621 (12) 0.07962 (17) 0.0226 (4)
C5 0.13959 (17) 0.86542 (13) 0.16921 (18) 0.0267 (4)
N 0.7271 (3) 0.5761 (2) 0.0020 (3) 0.0574 (6)
O1W 0.1645 (4) 0.5424 (5) 0.2555 (6) 0.177 (2)
O2W 0.0000 0.5920 (11) 0.7500 0.361 (10)
O3W 0.3966 (5) 0.2159 (4) 0.0572 (6) 0.0801 (16) 0.50
O3W′ 0.4277 (6) 0.1764 (5) −0.0637 (11) 0.137 (4) 0.50
O4W 0.4212 (7) 0.9401 (5) 0.012 (2) 0.517 (16)
O5W 0.4706 (12) 0.1707 (6) 0.1937 (10) 0.162 (5) 0.50
H1 0.302 (3) 0.748 (2) 0.212 (3) 0.050 (8)*
H11 0.774 (6) 0.630 (5) 0.020 (6) 0.15 (2)*
H12 0.741 (9) 0.520 (6) 0.044 (11) 0.22 (4)*
H13 0.668 (7) 0.587 (4) 0.046 (7) 0.16 (3)*
H14 0.695 (8) 0.573 (5) −0.088 (9) 0.23 (4)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Zn1 0.02137 (18) 0.01547 (17) 0.01684 (18) 0.000 0.00242 (11) 0.000
O1 0.0313 (7) 0.0220 (6) 0.0275 (7) 0.0057 (5) −0.0006 (6) 0.0048 (5)
O2 0.0272 (8) 0.0402 (9) 0.0424 (9) 0.0038 (6) −0.0003 (6) 0.0083 (7)
O3 0.0826 (14) 0.0575 (11) 0.0215 (8) 0.0381 (10) 0.0065 (8) −0.0012 (8)
O4 0.0384 (8) 0.0225 (7) 0.0272 (7) 0.0115 (5) 0.0081 (6) −0.0019 (5)
supporting information
sup-6
Acta Cryst. (2002). E58, m540–m542
C2 0.0261 (9) 0.0178 (8) 0.0224 (9) 0.0059 (6) 0.0005 (7) 0.0012 (7)
C3 0.0340 (10) 0.0248 (9) 0.0180 (8) 0.0087 (7) −0.0001 (7) −0.0007 (7)
C4 0.0281 (9) 0.0201 (8) 0.0198 (9) 0.0066 (7) 0.0038 (7) −0.0011 (6)
C5 0.0325 (10) 0.0256 (9) 0.0222 (9) 0.0077 (7) 0.0030 (7) −0.0042 (7)
N 0.0534 (15) 0.0608 (17) 0.0581 (17) −0.0100 (12) 0.0052 (13) −0.0016 (13)
O1W 0.096 (3) 0.241 (6) 0.197 (6) −0.005 (4) 0.052 (4) −0.027 (6)
O2W 0.300 (18) 0.292 (18) 0.48 (3) 0.000 −0.125 (18) 0.000
O3W 0.100 (4) 0.061 (3) 0.079 (4) −0.036 (3) −0.007 (3) 0.007 (3)
O3W′ 0.085 (5) 0.067 (4) 0.258 (13) −0.027 (3) 0.005 (6) 0.038 (6)
O4W 0.191 (9) 0.168 (7) 1.18 (5) −0.034 (6) −0.075 (16) 0.164 (16)
O5W 0.191 (12) 0.128 (6) 0.174 (12) −0.019 (7) 0.086 (11) −0.025 (6)
Geometric parameters (Å, º)
Zn1—O4i 1.9744 (14) C2—C4v 1.398 (3)
Zn1—O4ii 1.9744 (14) C3—C4 1.394 (3)
Zn1—O1iii 1.9789 (14) C3—H1 1.04 (3)
Zn1—O1 1.9789 (14) C4—C2v 1.398 (3)
O1—C1 1.276 (2) C4—C5 1.501 (2)
O2—C1 1.235 (3) N—H11 1.01 (7)
O3—C5 1.237 (3) N—H12 0.99 (10)
O4—C5 1.268 (2) N—H13 0.86 (8)
O4—Zn1iv 1.9744 (14) N—H14 1.05 (10)
C1—C2 1.507 (2) O3W—O3W′ 1.523 (12)
C2—C3 1.385 (3) O5W—O5Wiii 1.40 (2)
O4i—Zn1—O4ii 123.02 (9) C2—C3—H1 120.2 (17)
O4i—Zn1—O1iii 104.91 (6) C4—C3—H1 117.8 (17)
O4ii—Zn1—O1iii 103.79 (6) C3—C4—C2v 118.99 (17)
O4i—Zn1—O1 103.79 (6) C3—C4—C5 118.49 (17)
O4ii—Zn1—O1 104.91 (6) C2v—C4—C5 122.52 (16)
O1iii—Zn1—O1 117.40 (8) O3—C5—O4 123.50 (18)
C1—O1—Zn1 115.35 (12) O3—C5—C4 119.60 (17)
C5—O4—Zn1iv 112.59 (13) O4—C5—C4 116.89 (17)
O2—C1—O1 124.74 (17) H11—N—H12 124 (6)
O2—C1—C2 119.43 (17) H11—N—H13 99 (5)
O1—C1—C2 115.64 (17) H12—N—H13 91 (6)
C3—C2—C4v 119.46 (16) H11—N—H14 113 (6)
C3—C2—C1 116.62 (17) H12—N—H14 116 (7)
C4v—C2—C1 123.87 (16) H13—N—H14 107 (6)
C2—C3—C4 121.55 (18)