organic papers
Acta Cryst.(2006). E62, o347–o349 doi:10.1107/S1600536805042431 Dieltienset al. C
7H7NO2
o347
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
1-Methylpyrrole-2,5-dicarbaldehyde
Pieter E.M. Dieltiens,
Christophe M.L. Vande Velde, Herman J. Geise and
Frank Blockhuys*
Department of Chemistry, University of Antwerp, Universiteitsplein 1, B-2610 Wilrijk, Belgium
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 291 K
Mean(C–C) = 0.003 A˚
Rfactor = 0.038
wRfactor = 0.107 Data-to-parameter ratio = 7.0
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2006 International Union of Crystallography Printed in Great Britain – all rights reserved
The solid-state structure of the title compound, C7H7NO2, is
reported. The crystal packing is shaped mainly through carbonyl–carbonyl interactions on the one hand, and C— H n(O) hydrogen bonds and(CO) interactions on the other.
Comment
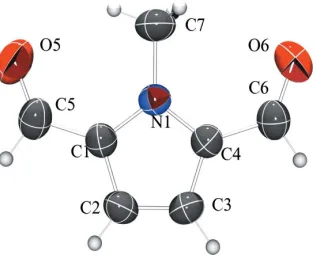
1-Methyl-2,5-pyrroledicarbaldehyde, (I) (Fig. 1), has been a sought-after intermediate, especially for the synthesis of organic semiconductor materials (Berlin et al., 1987; Cada-muroet al., 1993, 1996; Van Der Looyet al., 1997) but also for biologically active compounds and several macrocycles (Cadamuroet al., 1993, 1996).
The structure crystallizes in the non-centrosymmetric space group P212121 with no mirror symmetry present in the
mol-ecule or the structure. As a consequence, the two carbalde-hyde groups have different environments.
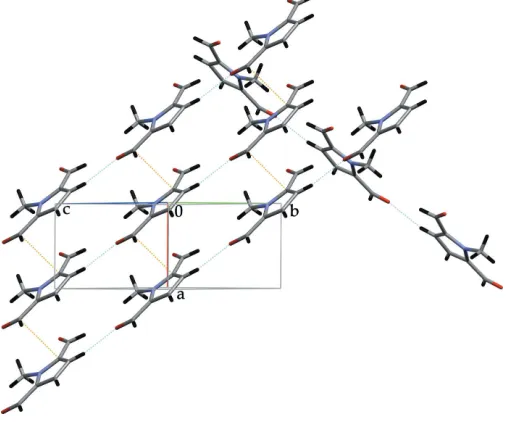
The crystal structure can be described as stacks of indivi-dual molecules along the [100] direction (Fig. 2) and by planes parallel to (001) consisting of stacked ribbons running along the [110] and [110] directions (Fig. 3). Between the stacks of individual molecules along the [100] direction, the stabilizing interactions are type II carbonyl–carbonyl interactions, as described by Allenet al.(1998). These are situated around the twofold screw axis and involve the carbonyl group C6 O6:
[image:1.610.267.396.335.409.2] [image:1.610.253.411.594.722.2]Received 14 December 2005 Accepted 19 December 2005 Online 23 December 2005
Figure 1
C6 O6i3.318 (3) A˚ [symmetry code (i)1 2+x,
1
2y,z] and
O6 C6i3.067 (3) A˚ . They are shown in Fig. 2 by the orange dotted lines. Perpendicular to the direction of these carbonyl– carbonyl contacts, the stacks display a short contact between the C atom of a methyl group and the O atom of a carbonyl group,viz. N1—C7 O6ii3.157 (3) A˚ , 179.06 (16)[symmetry code (ii)12+x,
1
2y,z]. This contact is shown in Fig. 2 by
the cyan dotted lines.
The remaining contacts involve the other carbonyl group C5 O5 and interact either within the aforementioned ribbons, such as the weak hydrogen bond C3—H3 O5iii 2.48 (3) A˚ , D A 3.417 (3) A˚ , 170 (2) [symmetry code (iii)
1 +x, 1 +y,z] (cyan dotted lines in Fig. 3), or between the ribbons, such as C4 C5iv 3.394 (4) A˚ [symmetry code (iv)
1 +x,y,z] (orange dotted lines in Fig. 3), forming the planes mentioned above. The latter contact is the expression of a typical(CO) contact, in this case given byCgA O5iv— C5iv3.598 (2) A˚ , 3.316 A˚ perp., 72.26 (17), whereCgA indi-cates the centroid of the pyrrole ring and ‘perp.’ indiindi-cates the perpendicular distance of O5 to the plane of the pyrrole ring. In conclusion, (I) displays a chiral structure which is completely dominated by interactions concerning the two different carbonyl groups: one of them is involved in carbonyl-carbonyl dipolar interactions, the other in(CO) contacts and CH n(O) hydrogen bonds. This is in contrast with the derivative which lacks the N-methyl group (Adams et al., 1986), which has an achiralPnstructure in which the crystal symmetry does not coincide with the molecular symmetry. Unsurprisingly, the network in the latter structure consists of hydrogen-bonded layers involving the H atoms of the alde-hyde group and the aromatic ring as well as those on the N atom. The O atoms of the carbonyl groups always act as acceptors. In addition, – interactions exist between the layers.
Experimental
All starting materials were obtained from Acros and used as received. Benzene was dried over 3 A molecular sieves prior to use.1H and13C
NMR spectra were recorded on a Varian Unity-400 apparatus. N-Methyl-2,5-pyrroledicarbaldehyde (I) was prepared starting fromN-methyl-2,5-bis(hydroxymethyl)pyrrole (II). For the synthesis of the latter N-methylpyrrole (22 ml, 0.255 mol), ground para-formaldehyde (16.1 g, 0.536 mol), potassium carbonate (0.2 g, 0.0015 mol) and water (5 ml) were mixed together and heated under a nitrogen atmosphere to 323–333 K until all the paraformaldehyde had dissolved. The reaction mixture was then cooled in a water bath until completion of the exothermic process (ca30 min.). The mixture was subsequently reheated to 353 K for 3 h. To remove water, N -methylpyrrole and N-methyl-2-(hydroxymethyl)pyrrole from the resulting crude reaction mixture, it was distilled in vacuo (1– 2 mmH g/323 K). The remaining impure precipitate was then recrystallized from propanol (10–15 ml) and washed with a small amount of chloroform. Overnight cooling of the filtrate yielded an additional fraction of (II). The yield was 22.7 g (65%) of an off-white powder [m.p. 386–387 K (uncorrected) in accordance with the literature (Chelintzev & Maksorov, 1916; Severin & Ipach, 1975)]. (II) should be refrigerated to prevent oxidation. 1H NMR (CDCl3,
400 MHz, TMS):3.71 (s, 3H, CH3), 4.60 (d, 4H,J= 2.6 Hz, CH2),
6.40 (s, 2H, H3 and H4), 7.26 (s, 2H, OH). 13C NMR (CDCl 3,
100 MHz, TMS):30.6 (CH3), 57.1 (CH2), 132 (C2 and C5), 107.7 (C3
and C4).
N-methyl-2,5-pyrroledicarbaldehyde (I) was prepared by refluxing a mixture consisting of (II) (7.7 g, 0.055 mol), dry benzene (300 ml) and precipitated active manganese dioxide (25.0 g, 0.287 mol) for 6 h and subsequently stirring at room temperature for 24 h. The precipitate was filtered off and washed carefully with 1,4-dioxane or tetrahydrofuran. The impure dark-brown crystals were sublimed under reduced pressure (typically for 6 h at 0.1 mmH g/338 K) (Severin & Ipach, 1975). (I) was collected in a yield of 3.0 g (40%) as light-yellow crystals [m.p. 370 K (uncorrected); lit. 368–369 K (Cresp & Sargent, 1972; Cresp & Sargent,1973; Severin & Ipach, 1975)]. The NMR data of (I) are identical to those of Loaderet al.(1982) [1H
NMR] and Cadamuroet al.(1993) [13C NMR].
Crystal data
C7H7NO2 Mr= 137.14
Orthorhombic,P212121
a= 4.650 (1) A˚
b= 6.457 (1) A˚
c= 22.386 (4) A˚
V= 672.1 (2) A˚3 Z= 4
Dx= 1.355 Mg m 3
MoKradiation Cell parameters from 25
reflections
= 5.7–19.7
= 0.10 mm1 T= 291 (2) K Prism, light yellow 0.40.30.2 mm
organic papers
o348
Dieltienset al. C [image:2.610.315.565.72.193.2] [image:2.610.314.567.229.440.2]7H7NO2 Acta Cryst.(2006). E62, o347–o349
Figure 2
View of the structure projected on to thebcplane. For details, see text.
Figure 3
Data collection
Enraf–Nonius Mach3 diffractometer
!/2scans
Absorption correction: none 1499 measured reflections 766 independent reflections 652 reflections withI> 2(I)
Rint= 0.032
max= 25.3 h= 0!5
k= 0!7
l=26!26 3 standard reflections
frequency: 60 min intensity decay: 21%
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.038 wR(F2) = 0.107 S= 1.00 766 reflections 109 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2(F
o2) + (0.0701P)2
+ 0.0615P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.16 e A˚
3
min=0.14 e A˚
3
The large value for the intensity decay was caused by the volatility of the compound. Sublimation took place during the course of the measurement. All H atoms were located in a difference density map and refined freely [C—H = 0.90 (3)–0.99 (3) A˚ ], except the methyl H atoms, for which a common distance was refined and the group was rotated so that it coincides with the maxima in the Fourier difference map [C—H = 0.92 A˚ andUiso(H) = 1.5Ueq(C)].
Data collection: CAD-4 EXPRESS (Enraf–Nonius, 1994); cell refinement: CAD-4 EXPRESS; data reduction:XCAD4 (Harms & Wocadlo, 1995); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure:SHELXL97 (Sheldrick, 1997); molecular graphics: MERCURY (Bruno et al., 2002) andORTEP-3 for Windows(Farrugia, 1997); software used to prepare material for publication:PLATON(Spek, 2003) andWinGX (Farrugia, 1999).
CVV thanks the FWO Vlaanderen for a grant as a Research Assistant. The authors thank Professor Dr. R. Dommisse and J. Aerts for the NMR measurements.
References
Adams, H., Beailey, N. A., Fenton, D. E., Moss, S, Rodriguez de Barbarin, C. O. & Jones, G. (1986).J. Chem. Soc. Dalton Trans.pp. 693–699.
Allen, F. H., Baalham, C. A., Lommerse J. P. M. & Raithby P. R. (1998).Acta Cryst.B54, 320–329.
Berlin, A., Bradamante, S., Ferracciolo, R., Pagani, G. A. & Sannicolo`, F. (1987).J. Chem. Soc. Perkin Trans. 1, pp. 2631–2635.
Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M. K., Macrae, C. F., McCabe, P., Pearson, J. & Taylor, R. (2002).Acta Cryst.B58, 389–397.
Cadamuro, S., Degani, I., Fochi, R., Gatti, A. & Piscopo, L. (1993).J. Chem. Soc. Perkin Trans. 1, pp. 2939–2944.
Cadamuro, S., Degani, I., Gatti, A. & Piscopo, L. J. (1996).J. Chem. Soc. Perkin Trans. 1, pp. 2365–2369.
Chelintzev, V. V. & Maksorov, B. V. (1916).J. Russ. Phys. Chem. Soc.48, 748– 779.
Cresp, T. M. & Sargent, M. V. (1972).J. Chem. Soc. Chem. Comm.pp. 807– 808.
Cresp, T. M. & Sargent, M. V. (1973).J. Chem. Soc. Perkin Trans. 1, pp. 2961– 2971.
Enraf–Nonius (1994).CAD-4 EXPRESS. Enraf–Nonius, Delft, The Nether-lands.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565. Farrugia, L. J. (1999).J. Appl. Cryst.32, 837–838.
Harms, K. & Wocadlo, S. (1995).XCAD4. University of Marburg, Germany. Loader, C. E., Barnett, G. H. & Anderson, H. J. (1982).Can. J. Chem.60, 383–
289.
Severin, Th. & Ipach, I. (1975).Chem. Ber.108, 1768–1775.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
Spek, A. L. (2003).J. Appl Cryst.36, 7–13.
Van Der Looy, J. F. A., Thys, G. J. H., Dieltiens, P. E. M., De Schrijver, D., Van Alsenoy, C. & Geise, H. J. (1997).Tetrahedron,53, 15069–15084.
organic papers
Acta Cryst.(2006). E62, o347–o349 Dieltienset al. C
supporting information
sup-1
Acta Cryst. (2006). E62, o347–o349supporting information
Acta Cryst. (2006). E62, o347–o349 [doi:10.1107/S1600536805042431]
1-Methylpyrrole-2,5-dicarbaldehyde
Pieter E.M. Dieltiens, Christophe M.L. Vande Velde, Herman J. Geise and Frank Blockhuys
S1. Comment
1-Methyl-2,5-pyrroledicarbaldehyde, (I) (Fig. 1), has been a sought after intermediate, especially for the synthesis of
organic semiconductor materials (Berlin et al., 1987; Cadamuro et al., 1993, 1996; Van Der Looy et al., 1997) but also
for biologically active compounds and several macrocycles (Cadamuro et al., 1993, 1996).
The structure crystallizes in the chiral space group P212121 with no mirror symmetry present in the molecule or the
structure. As a consequence, the two carbaldehyde moieties are surrounded in a different manner.
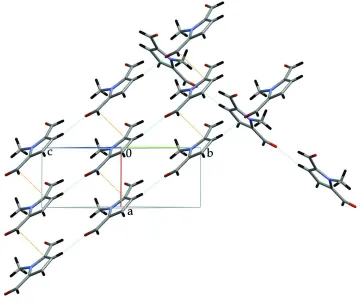
The crystal can be rationalized by stacks of individual molecules along the (100) direction (Fig. 2) and by planes in the
(001) direction consisting of stacked ribbons running along the (110) and (110) directions (Fig. 3). Between the stacks of
individual molecules along the (100) direction, the stabilizing interactions are type II carbonyl–carbonyl interactions, as
described by Allen et al. (1998). These are situated around the twofold screw axis and involve the carbonyl group
C6=O6: C6···O6i 3.318 (3) Å [symmetry code (i) −1/2 + x, 1/2 − y, −z] and O6)···C6i 3.067 (3) Å. They are shown in Fig.
2 by the orange dotted lines. Perpendicular to the direction of these carbonyl–carbonyl contacts, the stacks display a short
contact between the C atom of a methyl group and the O atom of a carbonyl group, viz. N1—C7···O6ii 3.157 (3) Å,
179.06 (16)° [symmetry code (ii) 1/2 + x, −1/2 − y, −z]. This contact is shown in Fig. 2 by the cyan dotted lines.
The remaining contacts involve the other carbonyl group C5=O5 and interact either within the aforementioned ribbons,
such as the weak hydrogen bond C3—H3···O5iii 2.48 (3) Å, D—A 3.417 (3) Å, 170 (2)° [symmetry code (iii) −1 + x, 1 +
y, z] (cyan dotted lines in Fig. 3), or inbetween the ribbons, such as C4···C5iv 3.394 (4) Å [symm. code (iv) −1 + x, y, z]
(orange dotted lines in Fig. 3), forming the planes mentioned above. The latter contact is the expression of a typical
π(CO)···π contact, in this case given by CgA···O5iv—C5iv 3.598 (2) Å, 3.316 Å perp., 72.26 (17)°, where CgA indicates
the centroid of the pyrrole ring and `perp.′ indicates the perpendicular distance of O5 to the plane of the pyrrole ring.
In conclusion, (I) displays a chiral structure which is completely dominated by interactions concerning the two different
carbonyl groups: one of them is involved in carbonyl-carbonyl dipolar interactions, the other in π(CO)···π contacts and
CH···n(O) hydrogen bonds. This is in contrast with the derivative which lacks the N-methyl group (Adams et al., 1986),
which has an achiral Pn structure in which the crystal symmetry does not coincide with the molecular symmetry.
Unsurprisingly, the network in the latter structure consists of hydrogen-bonded layers involving the H atoms of the
aldehyde group and the aromatic ring as well as those on the N atom. Yet, the On atoms of the carbonyl groups always act
as acceptors. In addition, π–π interactions exist between the layers.
S2. Experimental
All starting materials were obtained from Acros and used as received. Benzene was dried over 3 A molecular sieves prior
to use. 1H and 13C NMR spectra were recorded on a Varian Unity-400 apparatus.
N-methyl-2,5-pyrroledicarbaldehyde (I) was prepared starting from N-methyl-2,5-bis(hydroxymethyl)pyrrole (II). For
supporting information
sup-2
Acta Cryst. (2006). E62, o347–o349carbonate (0.2 g, 0.0015 mol) and water (5 ml) were mixed together and heated under a nitrogen atmosphere to 323–333
K until all the paraformaldehyde had dissolved. The reaction mixture was then cooled in a waterbath until completion of
the exothermic process (ca 30 min.). The mixture was subsequently reheated to 353 K for 3 h. To remove water, N
-methylpyrrole and N-methyl-2-(hydroxymethyl)pyrrole from the resulting crude reaction mixture, it was distilled in
vacuo (1–2 m mH g/323 K). The remaining impure precipitate was then recrystallized from propanol (10–15 ml) and
washed with a small amount of chloroform. Overnight cooling of the filtrate yielded an additional fraction of (II). The
yield was 22.7 g (65%) of an off-white powder [m.p. 386–387 K (uncorrected) in accordance with the literature
(Chelintzev & Maksorov, 1916; Severin & Ipach, 1975)]. (II) should be refrigerated to prevent oxidation. 1H NMR
(CDCl3, 400 MHz, TMS): δ 3.71 (s, 3H, CH3), 4.60 (d, 4H, J = 2.6 Hz, CH2), 6.40 (s, 2H, H3 and H4), 7.26 (s, 2H, OH).
13C NMR (CDCl
3, 100 MHz, TMS): δ 30.6 (CH3), 57.1 (CH2), 132 (C2 and C5), 107.7 (C3 and C4).
N-methyl-2,5-pyrroledicarbaldehyde (I) was prepared by refluxing a mixture consisting of (II) (7.7 g, 0.055 mol), dry
benzene (300 ml) and precipitated active manganese dioxide (25.0 g, 0.287 mol) for 6 h and subsequently stirring at room
temperature for 24 h. The precipitate was filtered off and washed carefully with 1,4-dioxane or tetrahydrofurane. The
impure dark-brown crystals were sublimed under reduced pressure (typically for 6 h at 0.1 m mH g/338 K) (Severin &
Ipach, 1975). (I) was collected in a yield of 3.0 g (40%) as light-yellow crystals [m.p. 370 K (uncorrected); lit. 368–369
K (Cresp & Sargent, 1972; Cresp & Sargent,1973; Severin & Ipach, 1975)]. The NMR data of (I) are identical to those of
Loader et al. (1982) [1H NMR] and Cadamuro et al. (1993) [13C NMR].
S3. Refinement
All H atoms were located in a difference density map and left free to refine [C—H = 0.90 (3)–0.99 (3) Å], except the
methyl H atoms, for which a common distance was refined and the group was twisted so that it coincides with the
supporting information
[image:6.610.127.481.72.361.2]sup-3
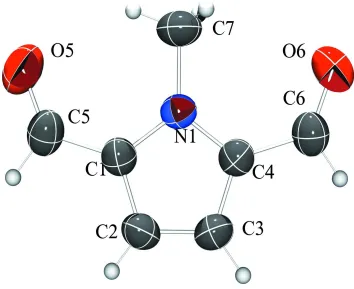
Acta Cryst. (2006). E62, o347–o349Figure 1
View of (1), including the numbering scheme. Displacement ellipsoids are drawn at the 50% probability level. H atoms
are numbered according to the C atom they are substituted on.
Figure 2
[image:6.610.127.484.415.596.2]supporting information
[image:7.610.125.488.71.375.2]sup-4
Acta Cryst. (2006). E62, o347–o349Figure 3
View of the structure along the bc cell diagonal. For details, see Comment.
1-Methylpyrrole-2,5-dicarbaldehyde
Crystal data
C7H7NO2
Mr = 137.14
Orthorhombic, P212121 Hall symbol: P 2ac 2ab a = 4.650 (1) Å b = 6.457 (1) Å c = 22.386 (4) Å V = 672.1 (2) Å3
Z = 4 F(000) = 288
Dx = 1.355 Mg m−3 Melting point: 370 K
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 25 reflections θ = 5.7–19.7°
µ = 0.10 mm−1
T = 291 K
Prism, light yellow 0.4 × 0.3 × 0.2 mm
Data collection
Enraf–Nonius Mach3 diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω/2θ scans
1499 measured reflections 766 independent reflections 652 reflections with I > 2σ(I)
Rint = 0.032
θmax = 25.3°, θmin = 1.8°
h = 0→5 k = 0→7 l = −26→26
supporting information
sup-5
Acta Cryst. (2006). E62, o347–o349Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.038
wR(F2) = 0.107
S = 1.00 766 reflections 109 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0701P)2 + 0.0615P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.16 e Å−3 Δρmin = −0.14 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O6 0.6125 (5) 0.0711 (3) 0.01315 (8) 0.0714 (6)
C4 0.8443 (5) 0.1555 (3) 0.10499 (9) 0.0454 (5)
N1 1.0208 (4) −0.0097 (2) 0.11338 (7) 0.0439 (5)
C1 1.1625 (5) 0.0190 (3) 0.16646 (9) 0.0461 (5)
C2 1.0708 (6) 0.2034 (4) 0.19095 (10) 0.0538 (6)
C6 0.6496 (6) 0.1870 (5) 0.05518 (10) 0.0575 (6)
C3 0.8729 (6) 0.2887 (4) 0.15297 (10) 0.0521 (6)
O5 1.4762 (5) −0.2716 (3) 0.17326 (10) 0.0828 (7)
C5 1.3801 (6) −0.1129 (5) 0.19272 (12) 0.0590 (7)
C7 1.0498 (7) −0.1891 (4) 0.07389 (11) 0.0633 (7)
H7A 0.872 (3) −0.246 (2) 0.0672 (7) 0.095*
H7B 1.167 (4) −0.285 (2) 0.0914 (5) 0.095*
H7C 1.128 (4) −0.1480 (10) 0.0383 (7) 0.095*
H3 0.781 (7) 0.420 (4) 0.1572 (11) 0.064 (8)*
H2 1.146 (6) 0.264 (4) 0.2235 (11) 0.049 (6)*
H6 0.566 (5) 0.322 (4) 0.0589 (9) 0.043 (6)*
H5 1.447 (7) −0.052 (5) 0.2312 (13) 0.077 (9)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O6 0.0777 (13) 0.0826 (13) 0.0540 (9) −0.0049 (12) −0.0106 (9) −0.0054 (9)
C4 0.0459 (12) 0.0476 (12) 0.0426 (11) 0.0004 (10) 0.0048 (10) 0.0028 (9)
supporting information
sup-6
Acta Cryst. (2006). E62, o347–o349C1 0.0427 (11) 0.0538 (13) 0.0417 (10) 0.0017 (11) 0.0042 (9) 0.0021 (9)
C2 0.0540 (14) 0.0604 (14) 0.0469 (12) −0.0008 (13) −0.0006 (11) −0.0112 (11)
C6 0.0597 (15) 0.0631 (16) 0.0497 (13) 0.0020 (15) −0.0014 (11) 0.0077 (12)
C3 0.0542 (14) 0.0487 (13) 0.0534 (12) 0.0044 (13) 0.0064 (11) −0.0044 (10)
O5 0.0874 (15) 0.0788 (13) 0.0824 (13) 0.0343 (13) 0.0044 (11) 0.0087 (11)
C5 0.0523 (14) 0.0669 (16) 0.0578 (15) 0.0105 (14) 0.0050 (13) 0.0104 (12)
C7 0.0773 (18) 0.0523 (13) 0.0602 (13) 0.0043 (15) 0.0059 (14) −0.0139 (12)
Geometric parameters (Å, º)
O6—C6 1.214 (3) C2—H2 0.90 (2)
C4—N1 1.359 (3) C6—H6 0.96 (2)
C4—C3 1.383 (3) C3—H3 0.96 (3)
C4—C6 1.451 (3) O5—C5 1.200 (3)
N1—C1 1.372 (3) C5—H5 1.00 (3)
N1—C7 1.463 (3) C7—H7A 0.9153
C1—C2 1.379 (3) C7—H7B 0.9153
C1—C5 1.447 (3) C7—H7C 0.9153
C2—C3 1.369 (4)
N1—C4—C3 108.85 (19) C4—C6—H6 108.3 (13)
N1—C4—C6 126.4 (2) C2—C3—C4 107.3 (2)
C3—C4—C6 124.7 (2) C2—C3—H3 126.7 (16)
C4—N1—C1 107.69 (17) C4—C3—H3 125.9 (16)
C4—N1—C7 126.42 (19) O5—C5—C1 128.0 (3)
C1—N1—C7 125.87 (19) O5—C5—H5 122.5 (17)
N1—C1—C2 108.2 (2) C1—C5—H5 109.6 (17)
N1—C1—C5 127.5 (2) N1—C7—H7A 109.5
C2—C1—C5 124.3 (2) N1—C7—H7B 109.5
C3—C2—C1 108.0 (2) H7A—C7—H7B 109.5
C3—C2—H2 126.3 (15) N1—C7—H7C 109.5
C1—C2—H2 125.3 (15) H7A—C7—H7C 109.5
O6—C6—C4 126.7 (3) H7B—C7—H7C 109.5
O6—C6—H6 124.8 (13)
C3—C4—N1—C1 −0.3 (2) C5—C1—C2—C3 177.7 (2)
C6—C4—N1—C1 −178.6 (2) N1—C4—C6—O6 0.2 (4)
C3—C4—N1—C7 178.2 (2) C3—C4—C6—O6 −177.8 (3)
C6—C4—N1—C7 0.0 (4) C1—C2—C3—C4 −0.1 (3)
C4—N1—C1—C2 0.2 (2) N1—C4—C3—C2 0.3 (3)
C7—N1—C1—C2 −178.3 (2) C6—C4—C3—C2 178.5 (2)
C4—N1—C1—C5 −177.5 (2) N1—C1—C5—O5 −0.3 (4)
C7—N1—C1—C5 4.0 (4) C2—C1—C5—O5 −177.7 (3)