organic papers
o3410
Kubo and Nakasuji C30H18 doi:10.1107/S1600536806022744 Acta Cryst.(2006). E62, o3410–o3411 Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
Bis(9-phenanthryl)ethyne
Takashi Kubo and Kazuhiro Nakasuji*
Department of Chemistry, Graduate School of Science, Osaka University, Toyonaka, Osaka 560-0043, Japan
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 150 K
Mean(C–C) = 0.002 A˚ Rfactor = 0.045 wRfactor = 0.137
Data-to-parameter ratio = 14.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 6 June 2006 Accepted 14 June 2006
#2006 International Union of Crystallography All rights reserved
The molecule of the title compound, C30H18, is located on an
inversion center and has an almost completely planar geometry.
Comment
Diarylethynes have been extensively investigated with respect to the wavelength–conjugation length relationship using UV spectroscopy (Akiyama et al., 1971; Nakagawa et al., 1971). Most of the compounds, however, have not been studied crystallographically, so the detailed structure–property rela-tionship is still open for discussion. We report here the crystal structure of bis(9-phenanthryl)ethyne, (I).
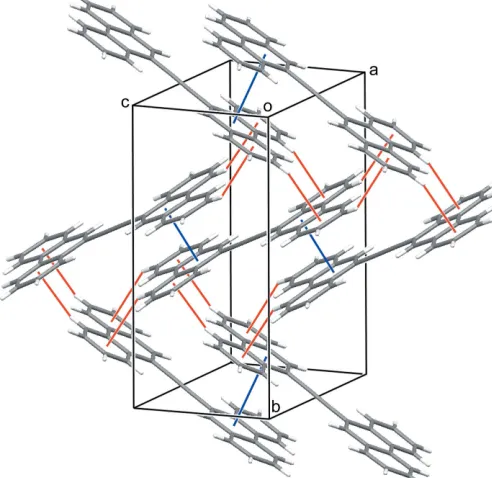
The molecule of (I), located an inversion center, has an almost completely planar geometry (Fig. 1). The bond lengths of the linker unit between the two phenanthrene rings, C1— C1(1x, y, 2z) and C1—C2 (Table 1), are typical for Csp—Csp and Csp—Csp2bonds, respectively (Bastiansen & Traetteberg, 1962). The molecules form a chain running along thecaxis through a–interaction between neighboring C2– C4/C9/C10/C15 rings. The distance between the centroids [Cg1 Cg1(1x,y, 1z)] is 3.7566 (6) A˚ . The chains are connected by C—H interactions (Fig. 2 and Table 2).
Experimental
The title compound was prepared according to the procedure of Mio et al.(2002). A Schlenk tube was charged with 9-bromophenanthrene (514 mg, 2.00 mmol), PdCl2(PPh3)2(42 mg, 0.060 mmol), CuI (38 mg,
0.20 mmol), 1,8-diazabicyclo[5.4.0]undec-7-ene (1.79 ml, 12.0 mmol), water (9.0ml, 0.50 mmol) and benzene (10 ml). Trimethylsilylacetyl-ene (0.14 ml, 1.0 mmol) was added to the mixture, which was degassed three times, and the reaction tube was purged with dry argon. The reaction tube was capped tightly and heated at 353 K for 2 d. After cooling, the reaction mixture was extracted with CH2Cl2,
washed with 0.5 N HCl (twice) and saturated aqueous NaCl, and dried over Na2SO4. The crude product was purified by silica gel
Crystal data
C30H18
Mr= 378.47
Monoclinic,P21=c
a= 9.3403 (8) A˚
b= 15.2266 (11) A˚
c= 6.9007 (5) A˚
= 102.0338 (13)
V= 959.86 (13) A˚3
Z= 2
Dx= 1.309 Mg m
3 MoKradiation
= 0.07 mm1
T= 150 (2) K Prism, colorless 0.450.300.20 mm
Data collection
Rigaku/MSC Mercury CCD diffractometer
!scans
Absorption correction: none 9274 measured reflections
2151 independent reflections 2036 reflections withI> 2(I)
Rint= 0.019
max= 27.5
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.045
wR(F2) = 0.137
S= 1.10 2151 reflections 145 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0872P)2 + 0.1528P]
whereP= (Fo 2
+ 2Fc 2
)/3 (/)max< 0.001
max= 0.33 e A˚
3
min=0.19 e A˚
3
Table 1
Selected geometric parameters (A˚ ,).
C1—C1i
1.197 (2) C1—C2 1.4351 (13)
C2—C3 1.3648 (14) C2—C15 1.4538 (13)
C1i
—C1—C2 178.37 (14)
Symmetry code: (i)xþ1;y;zþ2.
Table 2
Hydrogen-bond geometry (A˚ ,).
Cg2 andCg3 are the centroids of the C4–C9 and C10–C15 rings, respectively.
D—H A D—H H A D A D—H A
C7—H4 Cg2ii
0.95 2.94 3.6512 (12) 133 C11—H6 Cg3ii
0.95 2.82 3.5932 (11) 140
Symmetry code: (ii)x;yþ1 2;z
1 2.
All H atoms were placed in geometrically idealized positions (C— H = 0.95 A˚ ) and constrained to ride on their parent atoms, with Uiso(H) = 1.2Ueq(C).
Data collection:CrystalClear(Rigaku/MSC, 2002); cell refinement: CrystalClear; data reduction: TEXSAN (Rigaku/MSC, 2004); program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure:SHELXL97(Sheldrick, 1997); molecular graphics: ORTEP-3 (Farrugia, 1997); software used to prepare material for publication:TEXSAN.
This work is supported by Mitsubishi Chemical Corporation Fund and a Grant-in-Aid for Scientific Research on Priority Areas (No. 17550034) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
References
Akiyama, S., Nakasuji, K. & Nakagawa, M. (1971).Bull. Chem. Soc. Jpn,44, 2231–2236.
Bastiansen, O. & Traetteberg, M. (1962).Tetrahedron,17, 147–154. Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Mio, M. J., Kopel, L. C., Braun, J. C., Gadzikwa, T. L., Hull, K. L., Brisbois, R. G., Markworth, C. J. & Grieco, P. A. (2002).Org. Lett.4, 3199–3202. Nakagawa, M., Akiyama, S., Nakasuji, K. & Nishimoto, K. (1971).
Tetrahedron,27, 5401–5418.
Rigaku/MSC (2002).CrystalClear. Version 1.35. Rigaku/MSC, 9009 New Trails Drive, The Woodlands, TX 77381-5209, USA.
Rigaku/MSC. (2004).TEXSAN. Version 2.0. Rigaku/MSC, 9009 New Trails Drive, The Woodlands, TX 77381-5209, USA.
[image:2.610.316.566.73.217.2]Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
Figure 1
[image:2.610.315.561.269.508.2] [image:2.610.44.296.487.536.2]Drawing of (I), showing the atom-labeling scheme. Displacement ellipsoids are drawn at the 50% probability level. Unlabeled atoms are related to labeled atoms by 1x,y, 2z.
Figure 2
supporting information
sup-1 Acta Cryst. (2006). E62, o3410–o3411
supporting information
Acta Cryst. (2006). E62, o3410–o3411 [https://doi.org/10.1107/S1600536806022744]
Bis(9-phenanthryl)ethyne
Takashi Kubo and Kazuhiro Nakasuji
Bis(9-phenanthryl)ethyne
Crystal data
C30H18
Mr = 378.47
Monoclinic, P21/c Hall symbol: -P 2ybc
a = 9.3403 (8) Å
b = 15.2266 (11) Å
c = 6.9007 (5) Å
β = 102.0338 (13)°
V = 959.86 (13) Å3
Z = 2
F(000) = 396
Dx = 1.309 Mg m−3
Mo Kα radiation, λ = 0.7107 Å Cell parameters from 2216 reflections
θ = 4.0–27.5°
µ = 0.07 mm−1
T = 150 K Prism, colorless 0.45 × 0.30 × 0.20 mm
Data collection
Rigaku/MSC Mercury CCD diffractometer
Detector resolution: 14.63 pixels mm-1
ω scans
9274 measured reflections 2151 independent reflections
2036 reflections with I > 2σ(I)
Rint = 0.019
θmax = 27.5°, θmin = 4.0°
h = −12→12
k = −19→19
l = −8→8
Refinement
Refinement on F2
R[F2 > 2σ(F2)] = 0.045
wR(F2) = 0.137
S = 1.10 2151 reflections 145 parameters
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0872P)2 + 0.1528P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.33 e Å−3 Δρmin = −0.19 e Å−3
Special details
Experimental. 1H NMR (CDCl
3): δ 8.78–8.70 (m, 3H), 8.25 (s, 1H), 7.95 (dd, 1H, J = 7.6, 1.5 Hz), 7.80–7.62 (m, 4H); EI–MS (m/z, %): 378 (M+, 100).
Refinement. Refinement was carried out using all reflections. The weighted R-factor (wR) and goodness of fit (S) are based on F2. R-factor (gt) are based on F. The threshold expression of F2 > 2.0 σ(F2) is used only for calculating R-factor (gt).
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C1 0.47993 (11) 0.01799 (6) 0.92192 (15) 0.0240 (2)
C3 0.29146 (11) 0.06268 (6) 0.64007 (15) 0.0234 (3)
C4 0.24495 (11) 0.10142 (6) 0.44845 (15) 0.0213 (2)
C5 0.09541 (12) 0.10037 (7) 0.35211 (17) 0.0268 (3)
C6 0.05009 (12) 0.13640 (7) 0.16696 (18) 0.0314 (3)
C7 0.15306 (13) 0.17465 (7) 0.07087 (16) 0.0312 (3)
C8 0.29921 (12) 0.17623 (7) 0.16138 (16) 0.0268 (3)
C9 0.34933 (11) 0.13988 (6) 0.35194 (15) 0.0205 (2)
C10 0.50284 (11) 0.14027 (6) 0.45150 (15) 0.0201 (2)
C11 0.61247 (11) 0.17975 (6) 0.36754 (16) 0.0250 (3)
C12 0.75734 (12) 0.17803 (7) 0.46387 (17) 0.0284 (3)
C13 0.79952 (11) 0.13606 (7) 0.64737 (17) 0.0283 (3)
C14 0.69535 (11) 0.09771 (7) 0.73422 (15) 0.0246 (3)
C15 0.54608 (11) 0.09947 (6) 0.63996 (14) 0.0202 (2)
H1 0.2203 0.0380 0.7043 0.032 (3)*
H2 0.0254 0.0744 0.4165 0.041 (4)*
H3 −0.0506 0.1354 0.1042 0.046 (4)*
H4 0.1218 0.1996 −0.0572 0.044 (4)*
H5 0.3676 0.2023 0.0944 0.039 (4)*
H6 0.5859 0.2080 0.2424 0.033 (3)*
H7 0.8292 0.2056 0.4053 0.037 (4)*
H8 0.8998 0.1341 0.7117 0.041 (4)*
H9 0.7243 0.0697 0.8593 0.035 (3)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C1 0.0263 (5) 0.0253 (5) 0.0205 (5) 0.0005 (3) 0.0054 (4) 0.0019 (4) C2 0.0272 (5) 0.0199 (5) 0.0167 (5) 0.0001 (3) 0.0047 (4) 0.0023 (3) C3 0.0259 (5) 0.0242 (5) 0.0209 (5) 0.0021 (4) 0.0070 (4) 0.0006 (3) C4 0.0231 (5) 0.0199 (5) 0.0201 (5) −0.0001 (3) 0.0029 (4) 0.0014 (3) C5 0.0228 (5) 0.0286 (5) 0.0282 (6) 0.0017 (4) 0.0035 (4) −0.0009 (4) C6 0.0252 (5) 0.0352 (6) 0.0296 (6) 0.0015 (4) −0.0039 (4) 0.0008 (4) C7 0.0332 (6) 0.0338 (6) 0.0226 (5) 0.0060 (4) −0.0030 (4) 0.0016 (4) C8 0.0290 (6) 0.0274 (5) 0.0232 (6) 0.0053 (4) 0.0034 (4) −0.0001 (4) C9 0.0242 (5) 0.0178 (5) 0.0186 (5) −0.0007 (3) 0.0026 (4) 0.0014 (3) C10 0.0236 (5) 0.0170 (4) 0.0196 (5) −0.0006 (3) 0.0038 (4) 0.0009 (3) C11 0.0276 (5) 0.0234 (5) 0.0239 (5) 0.0032 (4) 0.0055 (4) −0.0001 (4) C12 0.0258 (5) 0.0280 (5) 0.0323 (6) 0.0019 (4) 0.0083 (4) −0.0027 (4) C13 0.0220 (5) 0.0308 (5) 0.0304 (6) −0.0027 (4) 0.0016 (4) 0.0001 (4) C14 0.0265 (5) 0.0256 (5) 0.0200 (5) −0.0007 (4) 0.0006 (4) 0.0021 (4) C15 0.0240 (5) 0.0179 (4) 0.0185 (5) −0.0013 (3) 0.0035 (4) 0.0013 (3)
Geometric parameters (Å, º)
C1—C1i 1.197 (2) C8—C9 1.4135 (14)
C1—C2 1.4351 (13) C8—H5 0.9500
C2—C3 1.3648 (14) C9—C10 1.4551 (14)
supporting information
sup-3 Acta Cryst. (2006). E62, o3410–o3411
C3—C4 1.4303 (14) C10—C15 1.4218 (13)
C3—H1 0.9500 C11—C12 1.3779 (15)
C4—C5 1.4163 (14) C11—H6 0.9500
C4—C9 1.4169 (14) C12—C13 1.3997 (15)
C5—C6 1.3737 (16) C12—H7 0.9500
C5—H2 0.9500 C13—C14 1.3748 (15)
C6—C7 1.4034 (17) C13—H8 0.9500
C6—H3 0.9500 C14—C15 1.4105 (14)
C7—C8 1.3779 (15) C14—H9 0.9500
C7—H4 0.9500
C1i—C1—C2 178.37 (14) C8—C9—C4 118.18 (9)
C3—C2—C1 120.62 (9) C8—C9—C10 122.70 (9)
C3—C2—C15 120.17 (9) C4—C9—C10 119.12 (9)
C1—C2—C15 119.20 (9) C11—C10—C15 118.00 (9)
C2—C3—C4 121.70 (9) C11—C10—C9 122.39 (9)
C2—C3—H1 119.1 C15—C10—C9 119.61 (9)
C4—C3—H1 119.1 C12—C11—C10 121.15 (9)
C5—C4—C9 119.46 (9) C12—C11—H6 119.4
C5—C4—C3 120.60 (9) C10—C11—H6 119.4
C9—C4—C3 119.93 (9) C11—C12—C13 120.53 (9)
C6—C5—C4 120.97 (10) C11—C12—H7 119.7
C6—C5—H2 119.5 C13—C12—H7 119.7
C4—C5—H2 119.5 C14—C13—C12 119.81 (9)
C5—C6—C7 119.77 (10) C14—C13—H8 120.1
C5—C6—H3 120.1 C12—C13—H8 120.1
C7—C6—H3 120.1 C13—C14—C15 120.81 (9)
C8—C7—C6 120.32 (10) C13—C14—H9 119.6
C8—C7—H4 119.8 C15—C14—H9 119.6
C6—C7—H4 119.8 C14—C15—C10 119.68 (9)
C7—C8—C9 121.30 (10) C14—C15—C2 120.90 (9)
C7—C8—H5 119.4 C10—C15—C2 119.42 (9)
C9—C8—H5 119.4
C1—C2—C3—C4 −177.60 (9) C8—C9—C10—C15 −177.57 (9)
C15—C2—C3—C4 1.56 (14) C4—C9—C10—C15 2.24 (13)
C2—C3—C4—C5 177.75 (9) C15—C10—C11—C12 0.73 (14)
C2—C3—C4—C9 −1.11 (14) C9—C10—C11—C12 −179.05 (9)
C9—C4—C5—C6 −0.27 (15) C10—C11—C12—C13 0.67 (15)
C3—C4—C5—C6 −179.13 (10) C11—C12—C13—C14 −1.30 (16)
C4—C5—C6—C7 0.21 (17) C12—C13—C14—C15 0.50 (15)
C5—C6—C7—C8 −0.03 (17) C13—C14—C15—C10 0.91 (15)
C6—C7—C8—C9 −0.10 (17) C13—C14—C15—C2 −178.99 (9)
C7—C8—C9—C4 0.04 (15) C11—C10—C15—C14 −1.50 (13)
C7—C8—C9—C10 179.85 (9) C9—C10—C15—C14 178.29 (8)
C5—C4—C9—C8 0.14 (14) C11—C10—C15—C2 178.40 (8)
C3—C4—C9—C8 179.01 (9) C9—C10—C15—C2 −1.82 (13)
C3—C4—C9—C10 −0.80 (13) C1—C2—C15—C14 −1.01 (14)
C8—C9—C10—C11 2.21 (14) C3—C2—C15—C10 −0.07 (14)
C4—C9—C10—C11 −177.98 (8) C1—C2—C15—C10 179.10 (8)
Symmetry code: (i) −x+1, −y, −z+2.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C7—H4···Cg2ii 0.95 2.94 3.6512 (12) 133
C11—H6···Cg3ii 0.95 2.82 3.5932 (11) 140