Acta Cryst.(2003). E59, o619±o621 DOI: 10.1107/S1600536803007268 Jones and Lozano C2HBr3O2
o619
organic papers
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
Tribromoacetic acid
Peter G. Jones* and Virginia Lozano
Institut fuÈr Anorganische und Analytische Chemie, Technische UniversitaÈt Braunschweig, Postfach 3329, 38023 Braunschweig, Germany
Correspondence e-mail: [email protected]
Key indicators Single-crystal X-ray study T= 133 K
Mean(C±C) = 0.003 AÊ Rfactor = 0.022 wRfactor = 0.053
Data-to-parameter ratio = 26.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2003 International Union of Crystallography Printed in Great Britain ± all rights reserved
The packing of the title compound, C2HBr3O2, involves
regions of hydrogen-bonded carboxylic acid dimers, in turn linked by two contacts of the type Br Br and one Br O.
Comment
We have begun to investigate the structures of tribromo-acetates and therefore decided to determine the structure of the parent acid, tribromoacetic acid. The structure of trichloroacetic acid has recently been reported by Rajagopalet al.(2003).
The molecular structure of tribromoacetic acid, (I), is presented in Fig. 1. Bond lengths and angles may be regarded as normal, in particular the CÐO bond lengths, which show no signs of the disorder sometimes observed in carboxylic acids (for a detailed discussion, see Wilson, 2002). Atom Br2 is approximately synperiplanar to O1.
More interesting features are observed in the packing. Apart from the classical `carboxylic acid dimer' formed by hydrogen bonding, there are three signi®cant contacts involving atom Br1; 3.6284 (5) AÊ to Br2(xÿ1
2, yÿ12, z),
3.6193 (5) AÊ to Br3(ÿxÿ1
2,y+12,ÿz+12), and 3.009 (2) AÊ to
the carbonyl atom O2(xÿ1
2,y+12,z).
The Br Br contacts are established as `type II' in the classi®cation of Pedireddi et al. (1994) by the angles at bromine; 101.25 (7)/167.92 (7) for the ®rst and 93.77 (7)/ 167.05 (7) for the second. Such contacts are thought to be associated with a positive region of charge in the extension of the CÐBr vector beyond Br, which can interact with the negative region perpendicular to the CÐBr bond at the other bromine. The somewhat longer Br3 Br3 interaction of 3.8020 (5) AÊ (code:ÿxÿ1
2,yÿ12,ÿz+12), however, has equal
angles at bromine [142.57 (7), equal by symmetry] and such `type I' contacts are regarded as less energetically favourable. Contacts of the type Br O may be regarded as one form of `halogen bond' (Metrangolo & Resnati, 2001). Although the distance observed here is signi®cantly shorter than the sum of the van der Waals radii (Br = 1.85 AÊ and O = 1.52 AÊ; Bondi, 1964), much shorter values have been observed (down toca
2.7 AÊ, as quoted in the above-mentioned article). Lommerseet al. (1996), in a review of contacts between halogens and oxygen or nitrogen, pointed out that these also tend to be
organic papers
o620
Jones and Lozano C2HBr3O2 Acta Cryst.(2003). E59, o619±o621approximately linear at the halogen [here 157.24 (8)], but show little preferred directionality to the O-atom lone pairs [here the bromine lies 1.964 (5) AÊ out of the C1/C2/O1/O2 plane]. Again, electrostatic forces are thought to play an important role.
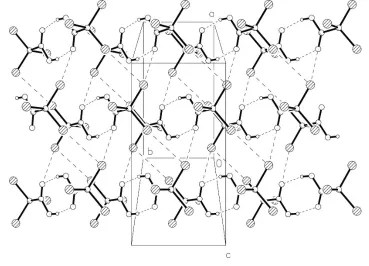
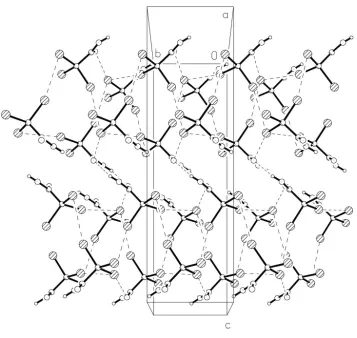
The net effect of the contacts can be seen in the packing diagrams. Carboxylic acid dimers occupy the regionsz'0,1
2,
1,etc. Fig. 2 shows the region atz'1
2, with dimer formation
supported by Br1 Br2 and Br1 O2; Br1 Br3 links the CBr3 moieties of successive dimer regions to form double
layers of molecules betweene.g. z'1
2and 1 (Fig. 3).
Experimental
The title material was purchased from Aldrich. Single crystals grew from a solution in dichloromethane/petrol ether (ca 1/5 v/v) on cooling to 255 K.
Crystal data
C2HBr3O2
Mr= 296.76
Monoclinic,C2=c a= 11.1679 (12) AÊ b= 5.7966 (6) AÊ c= 19.821 (2) AÊ = 103.132 (4)
V= 1249.6 (2) AÊ3
Z= 8
Dx= 3.155 Mg mÿ3
MoKradiation Cell parameters from 4624
re¯ections = 3.8±30.5
= 19.26 mmÿ1
T= 133 (2) K Tablet, colourless 0.150.130.04 mm
Data collection
Bruker SMART 1000CCD diffractometer !and'scans
Absorption correction: multi-scan (SADABS; Bruker, 1998) Tmin= 0.268,Tmax= 0.564
9331 measured re¯ections
1823 independent re¯ections 1577 re¯ections withI> 2(I) Rint= 0.037
max= 30.0
h=ÿ15!15 k=ÿ8!8 l=ÿ27!27
Re®nement
Re®nement onF2
R[F2> 2(F2)] = 0.022
wR(F2) = 0.053
S= 1.00 1823 re¯ections 68 parameters
All H-atom parameters re®ned w= 1/[2(F
o2) + (0.031P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001
max= 0.61 e AÊÿ3
min=ÿ0.80 e AÊÿ3
Table 1
Selected geometric parameters (AÊ,).
Br1ÐC1 1.934 (2) Br2ÐC1 1.929 (3) Br3ÐC1 1.944 (2)
O1ÐC2 1.302 (3) O2ÐC2 1.219 (3) C1ÐC2 1.539 (3)
C2ÐC1ÐBr2 110.56 (17) C2ÐC1ÐBr1 111.01 (17) Br2ÐC1ÐBr1 109.85 (12) C2ÐC1ÐBr3 105.38 (16) Br2ÐC1ÐBr3 110.08 (12)
Br1ÐC1ÐBr3 109.90 (12) O2ÐC2ÐO1 126.2 (2) O2ÐC2ÐC1 121.0 (2) O1ÐC2ÐC1 112.7 (2)
Br2ÐC1ÐC2ÐO2 ÿ16.4 (3)
Table 2
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A O1ÐH01 O2i 0.76 (4) 1.95 (4) 2.691 (3) 162 (4)
Symmetry code: (i)ÿx;ÿy;1ÿz.
Data collection:SMART(Bruker, 1998); cell re®nement:SAINT (Bruker, 1998); data reduction: SAINT; program(s) used to solve Figure 2
Packing diagram of the title compound viewed perpendicular to theab
plane. Secondary interactions are indicated by dashed lines.
Figure 3
Packing diagram of the title compound viewed perpendicular to thebc
plane. Secondary interactions are indicated by dashed lines. Figure 1
structure:SHELXS97 (Sheldrick, 1990); program(s) used to re®ne structure: SHELXL97 (Sheldrick, 1997); molecular graphics: XP (Siemens, 1994); software used to prepare material for publication: SHELXL97.
Financial support from the Fonds der Chemischen Industrie is gratefully acknowledged. VL was supported by the Erasmus scheme. We thank Mr A. Weinkauf for technical assistance.
References
Bondi, A. (1964).J. Phys. Chem.68, 441±451.
Bruker (1998).SMART(Version 5.0),SAINT(Version 4.0) andSADABS (Version 2.0). Bruker AXS Inc., Madison, Wisconsin, USA.
Lommerse, J. P. M., Stone, A. J., Taylor, R. & Allen, F. H. (1996).J. Am. Chem. Soc.118, 3108±3116.
Metrangolo, P. & Resnati, G. (2001).Chem. Eur. J.7, 2511±2519.
Pedireddi, V. R., Reddy, D. S., Goud, B. S., Craig, D. C., Rae, A. D. & Desiraju, G. R. (1994).J. Chem. Soc. Perkin Trans.2, pp. 2353±2360.
Rajagopal, K., Mostad, A., Krishnakumar, R. V., Nandhini, M. S. & Natarajan, S. (2003).Acta Cryst.E59, o316±o318.
Sheldrick, G. M. (1990).Acta Cryst.A46, 467±473.
Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany. Siemens (1994).XP. Version 5.03. Siemens Analytical X-ray Instruments Inc.,
Madison, Wisconsin, USA.
Wilson, C. C. (2002).New J. Chem.26, 1733±1739.
Acta Cryst.(2003). E59, o619±o621 Jones and Lozano C2HBr3O2
o621
supporting information
sup-1
Acta Cryst. (2003). E59, o619–o621
supporting information
Acta Cryst. (2003). E59, o619–o621 [doi:10.1107/S1600536803007268]
Tribromoacetic acid
Peter G. Jones and Virginia Lozano
S1. Comment
We have begun to investigate the structures of tribromoacetates and therefore decided to determine the structure of the
parent acid, tribromoacetic acid. The structure of trichloracetic acid has recently been reported by Rajagopal et al. (2003).
The molecular structure of tribromoacetic acid, (I), is presented in Fig. 1. Bond lengths and angles may be regarded as
normal, in particular the C—O bond lengths, which show no signs of the disorder sometimes observed in carboxylic
acids (for a detailed discussion, see Wilson, 2002). Atom Br2 is approximately synperiplanar to O1.
More interesting features are observed in the packing. Apart from the classical `carboxylic acid dimer′ formed by
hydrogen bonding, there are three significant contacts involving atom Br1; 3.6284 (5) Å to Br2(x − 1/2, y − 1/2, z),
3.6193 (5) Å to Br3(-x − 1/2, y + 1/2, −z + 1/2), and 3.009 (2) Å to the carbonyl atom O2(x − 1/2, y + 1/2, z).
The Br···Br contacts are established as `type II′ in the classification of Pedireddi et al. (1994) by the angles at bromine;
101.25 (7)/167.92 (7)° for the first and 93.77 (7)/167.05 (7)° for the second. Such contacts are thought to be associated
with a positive region of charge in the extension of the C—Br vector beyond Br, which can interact with the negative
region perpendicular to the C—Br bond at the other bromine. The somewhat longer Br3···Br3 interaction of 3.8020 (5) Å
(code: −x − 1/2, y − 1/2, −z + 1/2), however, has equal angles at bromine [142.57 (7)°, equal by symmetry] and such `type
I′ contacts are regarded as less energetically favourable.
Contacts of the type Br···O may be regarded as one form of `halogen bond′ (Metrangolo & Resnati, 2001). Although the
distance observed here is significantly shorter than the sum of the van der Waals radii (Br = 1.85 Å and O = 1.52 Å;
Bondi, 1964), much shorter values have been observed (down to ca 2.7 Å, as quoted in the above-mentioned article).
Lommerse et al. (1996), in a review of contacts between halogens and oxygen or nitrogen, pointed out that these also
tend to be approximately linear at the halogen [here 157.24 (8)°], but show little preferred directionality to the O-atom
lone pairs [here the bromine lies 1.964 (5) Å out of the C1/C2/O1/O2 plane]. Again, electrostatic forces are thought to
play an important role.
The net effect of the contacts can be seen in the packing diagrams. Carboxylic acid dimers occupy the regions z≈ 0,
1/2, 1, etc. Fig. 2 shows the region at z≈ 1/2, with dimer formation supported by Br1···Br2 and Br1···O2; Br1···Br3 links
the CBr3 moieties of successive dimer regions to form double layers of molecules between e.g. z≈ 1/2 and 1 (Fig. 3).
S2. Experimental
The title material was purchased from Aldrich. Single crystals grew from a solution in dichloromethane/petrol ether (ca
supporting information
sup-2
[image:5.610.126.485.78.348.2]Acta Cryst. (2003). E59, o619–o621 Figure 1
The molecule of the title compound in the crystal. Ellipsoids represent 50% probability levels. H-atom radii are arbitrary.
Figure 2
Packing diagram of the title compound viewed perpendicular to the xy plane. Secondary interactions are indicated by
[image:5.610.120.489.389.655.2]supporting information
sup-3
[image:6.610.128.485.66.404.2]Acta Cryst. (2003). E59, o619–o621 Figure 3
Packing diagram of the title compound viewed perpendicular to the yz plane. Secondary interactions are indicated by
dashed lines.
Tribromoacetic acid
Crystal data
C2HBr3O2
Mr = 296.76 Monoclinic, C2/c a = 11.1679 (12) Å b = 5.7966 (6) Å c = 19.821 (2) Å β = 103.132 (4)° V = 1249.6 (2) Å3
Z = 8
F(000) = 1072 Dx = 3.155 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 4624 reflections θ = 3.8–30.5°
µ = 19.26 mm−1
T = 133 K Tablet, colourless 0.15 × 0.13 × 0.04 mm
Data collection
Bruker SMART 1000 CCD diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 8.192 pixels mm-1
ω and φ scans
supporting information
sup-4
Acta Cryst. (2003). E59, o619–o621 θmax = 30.0°, θmin = 2.1°
h = −15→15
k = −8→8 l = −27→27
Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.022
wR(F2) = 0.053
S = 1.00 1823 reflections 68 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
All H-atom parameters refined w = 1/[σ2(F
o2) + (0.031P)2] where P = (Fo2 + 2Fc2)/3 (Δ/σ)max = 0.001
Δρmax = 0.61 e Å−3 Δρmin = −0.80 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Non-bonded contacts: Distances
3.6284 (0.0005) Br1 - Br2_$2 3.6193 (0.0005) Br1 - Br3_$3 3.8020 (0.0005) Br3 - Br3_$4 3.0091 (0.0020) Br1 - O2_$5 3.4620 (0.0022) Br1 - O1_$6
Angles
101.25 (0.07) C1 - Br1 - Br2_$2 167.92 (0.07) Br1 - Br2_$2 - C1_$2 93.77 (0.07) C1 - Br1 - Br3_$3 161.05 (0.07) Br1 - Br3_$3 - C1_$3 142.57 (0.07) C1 - Br3 - Br3_$4 157.24 (0.08) C1 - Br1 - O2_$5 129.94 (0.17) Br1 - O2_$5 - C2_$5 108.12 (0.08) C1 - Br1 - O1_$6 124.07 (0.16) Br1 - O1_$6 - C2_$6
Operators for generating equivalent atoms:
$2 x − 1/2, y − 1/2, z $3 − x − 1/2, y + 1/2, −z + 1/2 $4 − x − 1/2, y − 1/2, −z + 1/2 $5 x − 1/2, y + 1/2, z $6 x, y + 1, z Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Br1 −0.24614 (2) 0.57970 (4) 0.389915 (13) 0.01850 (7)
Br2 0.02550 (3) 0.62438 (5) 0.369499 (15) 0.02254 (8)
Br3 −0.14928 (3) 0.22989 (5) 0.291161 (13) 0.02456 (8)
O1 −0.14986 (18) 0.1241 (4) 0.45092 (11) 0.0219 (4)
H01 −0.134 (4) 0.014 (7) 0.472 (2) 0.046 (13)*
O2 0.04944 (17) 0.2079 (3) 0.45893 (10) 0.0213 (4)
C1 −0.1044 (2) 0.4116 (4) 0.37556 (12) 0.0148 (5)
C2 −0.0599 (2) 0.2358 (4) 0.43403 (12) 0.0155 (5)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Br1 0.01605 (13) 0.01810 (13) 0.02177 (13) 0.00315 (9) 0.00519 (10) 0.00137 (9)
Br2 0.01902 (15) 0.02053 (14) 0.02938 (15) −0.00299 (9) 0.00825 (11) 0.00536 (10)
supporting information
sup-5
Acta Cryst. (2003). E59, o619–o621
O1 0.0154 (10) 0.0233 (11) 0.0261 (10) −0.0013 (8) 0.0029 (8) 0.0101 (8)
O2 0.0155 (10) 0.0257 (10) 0.0226 (9) 0.0015 (8) 0.0042 (8) 0.0081 (8)
C1 0.0143 (12) 0.0155 (12) 0.0146 (11) 0.0000 (9) 0.0033 (9) 0.0010 (9)
C2 0.0185 (13) 0.0161 (12) 0.0123 (11) 0.0000 (9) 0.0041 (9) −0.0010 (9)
Geometric parameters (Å, º)
Br1—C1 1.934 (2) O2—C2 1.219 (3)
Br2—C1 1.929 (3) C1—C2 1.539 (3)
Br3—C1 1.944 (2) O1—H01 0.76 (4)
O1—C2 1.302 (3)
C2—C1—Br2 110.56 (17) Br1—C1—Br3 109.90 (12)
C2—C1—Br1 111.01 (17) O2—C2—O1 126.2 (2)
Br2—C1—Br1 109.85 (12) O2—C2—C1 121.0 (2)
C2—C1—Br3 105.38 (16) O1—C2—C1 112.7 (2)
Br2—C1—Br3 110.08 (12) C2—O1—H01 118 (3)
Br2—C1—C2—O2 −16.4 (3) Br2—C1—C2—O1 165.00 (18)
Br1—C1—C2—O2 −138.6 (2) Br1—C1—C2—O1 42.8 (3)
Br3—C1—C2—O2 102.5 (2) Br3—C1—C2—O1 −76.1 (2)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O1—H01···O2i 0.76 (4) 1.95 (4) 2.691 (3) 162 (4)