organic papers
Acta Cryst.(2006). E62, o2397–o2398 doi:10.1107/S1600536806017946 Patilet al. C
13H9NO4
o2397
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
3-(2-Furyl)-1-(4-nitrophenyl)prop-2-en-1-one
P. S. Patil,aJeannie Bee-Jan Teh,b Hoong-Kun Fun,b*
Ibrahim Abdul Razakb and S. M. Dharmaprakasha
aDepartment of Studies in Physics, Mangalore
University, Mangalagangotri, Mangalore 574 199, India, andbX-ray Crystallography Unit, School of Physics, Universiti Sains Malaysia, 11800 USM, Penang, Malaysia
Correspondence e-mail: hkfun@usm.my
Key indicators
Single-crystal X-ray study T= 100 K
Mean(C–C) = 0.002 A˚ Rfactor = 0.070 wRfactor = 0.198
Data-to-parameter ratio = 35.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 4 May 2006 Accepted 15 May 2006
#2006 International Union of Crystallography All rights reserved
In the title compound, C13H9NO4, the dihedral angle between
the benzene and furan rings is 6.23 (5). The crystal structure
is stabilized by weak intermolecular C—H O hydrogen bonds.
Comment
Chalcones show interesting biological and pharmacological activities (De Vincenzo et al., 1995; Kumar et al., 2003). In addition, chalcones with appropriate subtituents constitute a class of non-linear optical materials (Fichou et al., 1988; Kitaokaet al., 1990; Uchidaet al., 1998; Gotoet al., 1991, Patil et al., 2006a,b; Zhanget al., 1990; Zhao et al., 2000). In this paper, we present the crystal structure of the title compound, (I), which does not exhibit second-order non-linear optical properties as it crystallizes out in a centrosymmetric space group.
In (I) (Fig. 1), the bond lengths and angles have normal values (Allen et al., 1987), comparable with those found in related structures (Teh et al., 2006; Patilet al., 2006a,b; Nget al., 2006; Rosliet al., 2006). The least-squares plane through the enone group makes dihedral angles of 4.19 (5) and 2.14 (6), respectively, with the benzene and furan rings. The dihedral angle between the two rings is 6.23 (5). The nitro group at C3 is almost coplanar with the C1–C6 benzene ring, with O4—N1—C3—C4 and O3—N1—C3—C2 torsion angles of3.49 (19) and4.38 (18), respectively.
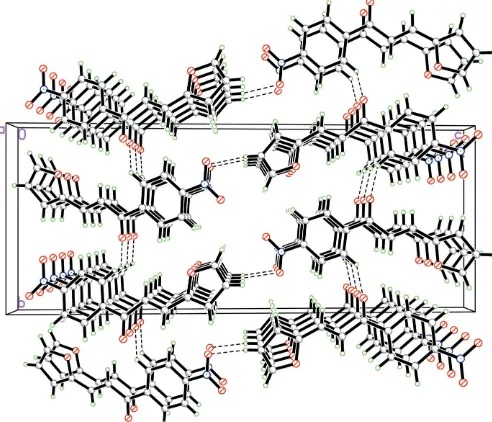
The crystal packing (Fig. 2) is stabilized by weak inter-molecular C—H O hydrogen bonds (Table 1).
Experimental
Crystal data
C13H9NO4
Mr= 243.21
Monoclinic,P21=c
a= 3.8809 (2) A˚
b= 10.6603 (4) A˚
c= 26.5500 (11) A˚
= 94.867 (3)
V= 1094.45 (8) A˚3
Z= 4
Dx= 1.476 Mg m3
MoKradiation
= 0.11 mm1
T= 100.0 (1) K Plate, yellow
0.540.380.10 mm
Data collection
Bruker SMART APEX2 CCD area-detector diffractometer
!scans
Absorption correction: multi-scan (SADABS; Bruker, 2005)
Tmin= 0.806,Tmax= 0.989
28259 measured reflections 5741 independent reflections 3956 reflections withI> 2(I)
Rint= 0.067
max= 37.5
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.070
wR(F2) = 0.198
S= 1.10 5741 reflections 163 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0956P)2
+ 0.1328P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.56 e A˚3
min=0.27 e A˚3
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
C1—H1A O2i
0.93 2.48 3.151 (2) 129 C12—H12A O3ii
0.93 2.55 3.463 (2) 169
Symmetry codes: (i)x;y1 2;zþ
1
2; (ii)x2;yþ 1 2;z
1 2.
H atoms were placed in calculated positions, with C—H distances of 0.93 A˚ , and treated as riding. TheUiso(H) values were constrained to be 1.2Ueq(C).
Data collection:APEX2(Bruker, 2005); cell refinement:APEX2; data reduction: SAINT (Bruker, 2005); program(s) used to solve structure: SHELXTL (Sheldrick, 1998); program(s) used to refine structure:SHELXTL; molecular graphics:SHELXTL; software used to prepare material for publication: SHELXTL,PARST(Nardelli, 1995) andPLATON(Spek, 2003).
The authors thank the Malaysian Government and Universiti Sains Malaysia for Scientific Advancement Grant Allocation (SAGA) grant No. 304/PFIZIK/653003/A118 and the USM short-term grant No. 304/PFIZIK/635028. PSP and SMD are grateful to DRDO, Government of India, for financial assistance.
References
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987).J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
Bruker (2005).APEX2(Version 1.27),SAINT(Version 7.12A) andSADABS
(Version 2004/1). Bruker AXS Inc., Madison, Wisconsin, USA.
De Vincenzo, R., Scambia, G., Benedetti, P. P., Ranelletti, F. O., Bonanno, G., Ercoli, A., Delle, M. F., Ferrari, F., Piantelli, M. & Mancuso, S. (1995).
Anticancer Drug Des.10, 481–490.
Fichou, D., Watanabe, T., Takeda, T., Miyata, S., Goto, Y. & Nakayama, M. (1988).Jpn J. Appl. Phys.27, L429–L430.
Goto, Y., Hayashi, A., Kimura, Y. & Nakayama, M. (1991).J. Cryst. Growth,
108, 688–698.
Kitaoka, Y., Sasaki, T., Nakai, S., Yokotani, A., Goto, Y. & Nakayama, M. (1990).Appl. Phys. Lett.56, 2074–2076.
Kumar, S. K., Hager, E., Pettit, C., Gurulingappa, H., Davidson, N. E. & Khan, S. R. (2003).J. Med. Chem.46, 2813–2815.
Nardelli, M. (1995).J. Appl. Cryst.28, 659.
Ng, S. L., Shettigar, V., Razak, I. A., Fun, H.-K., Patil, P. S. & Dharmaprakash, S. M. (2006).Acta Cryst.E62, o1421–o1423.
Patil, P. S., Teh, J. B. J., Fun, H.-K., Razak, I. A. & Dharmaprakash, S. M. (2006a).Acta Cryst.E62, o896–o898.
Patil, P. S., Teh, J. B. J., Fun, H.-K., Razak, I. A. & Dharmaprakash, S. M. (2006b).Acta Cryst.E62, o1710–o1712.
Rosli, M. M., Patil, P. S., Fun, H.-K., Razak, I. A., Dharmaprakash, S. M. & Karthikeyan, M. S. (2006).Acta Cryst.E62, o1460–o1462.
Sheldrick, G. M. (1998).SHELXTL. Version 5.10. Bruker AXS Inc., Madison, Wisconsin, USA.
Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
Teh, J. B. J., Patil, P. S., Fun, H.-K., Razak, I. A., Shettigar, V. & Dharmaprakash, S. M. (2006).Acta Cryst.E62, o1526–o1528.
Uchida, T., Kozawa, K., Sakai, T., Aoki, M., Yoguchi, H., Abdureyim, A. & Watanabe, Y. (1998).Mol. Cryst. Liq. Cryst.314, 135–140.
Zhang, G., Kinoshita, T., Sasaki, K., Goto, Y. & Nakayam, M. (1990).J. Cryst. Growth,100, 411–416.
[image:2.610.314.564.71.168.2]Zhao, B., Lu, W.-Q., Zhou, Z.-H. & Wu, Y. (2000).J. Mater. Chem.10, 1513– 1517.
Figure 1
[image:2.610.316.562.221.432.2]View of (I), showing the atomic numbering and 50% probability displacement ellipsoids.
Figure 2
The crystal packing, viewed down theaaxis. Hydrogen bonds are shown
supporting information
sup-1 Acta Cryst. (2006). E62, o2397–o2398
supporting information
Acta Cryst. (2006). E62, o2397–o2398 [https://doi.org/10.1107/S1600536806017946]
3-(2-Furyl)-1-(4-nitrophenyl)prop-2-en-1-one
P. S. Patil, Jeannie Bee-Jan Teh, Hoong-Kun Fun, Ibrahim Abdul Razak and S. M. Dharmaprakash
3-(2-Furyl)-1-(4-nitrophenyl)prop-2-en-1-one
Crystal data
C13H9NO4 Mr = 243.21 Monoclinic, P21/c Hall symbol: -P 2ybc
a = 3.8809 (2) Å
b = 10.6603 (4) Å
c = 26.5500 (11) Å
β = 94.867 (3)°
V = 1094.45 (8) Å3
Z = 4
F(000) = 504
Dx = 1.476 Mg m−3
Mo Kα radiation, λ = 0.71073 Å
Cell parameters from 4832 reflections
θ = 1.5–37.5°
µ = 0.11 mm−1
T = 100 K
Block, yellow
0.54 × 0.38 × 0.10 mm
Data collection
Bruker SMART APEX2 CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 8.33 pixels mm-1
ω scans
Absorption correction: multi-scan (SADABS; Bruker, 2005)
Tmin = 0.806, Tmax = 0.989
28259 measured reflections 5741 independent reflections 3956 reflections with I > 2σ(I)
Rint = 0.067
θmax = 37.5°, θmin = 1.5°
h = −6→6
k = −17→17
l = −45→45
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.070 wR(F2) = 0.198
S = 1.10
5741 reflections 163 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0956P)2 + 0.1328P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.56 e Å−3 Δρmin = −0.27 e Å−3
Special details
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 −0.1250 (3) 0.26693 (9) 0.10568 (4) 0.0231 (2)
O2 0.0305 (3) 0.59893 (8) 0.24281 (4) 0.0237 (2)
O3 1.0335 (3) 0.21435 (10) 0.42360 (4) 0.0336 (3)
O4 1.0389 (3) 0.40418 (10) 0.45266 (4) 0.0331 (3)
N1 0.9515 (3) 0.32556 (11) 0.42030 (4) 0.0222 (2)
C1 0.4735 (3) 0.31959 (11) 0.29253 (5) 0.0180 (2)
H1A 0.4232 0.2633 0.2662 0.022*
C2 0.6653 (3) 0.28027 (11) 0.33638 (5) 0.0187 (2)
H2A 0.7414 0.1978 0.3399 0.022*
C3 0.7400 (3) 0.36659 (11) 0.37458 (4) 0.0181 (2)
C4 0.6322 (3) 0.49069 (12) 0.37111 (5) 0.0208 (2)
H4A 0.6885 0.5470 0.3973 0.025*
C5 0.4379 (3) 0.52820 (11) 0.32744 (5) 0.0203 (2)
H5A 0.3609 0.6107 0.3243 0.024*
C6 0.3562 (3) 0.44321 (10) 0.28801 (4) 0.0160 (2)
C7 0.1394 (3) 0.49038 (11) 0.24248 (4) 0.0173 (2)
C8 0.0602 (3) 0.40802 (11) 0.19861 (4) 0.0190 (2)
H8A 0.1464 0.3266 0.1986 0.023*
C9 −0.1393 (3) 0.45199 (11) 0.15827 (5) 0.0194 (2)
H9A −0.2211 0.5335 0.1608 0.023*
C10 −0.2384 (3) 0.38741 (11) 0.11227 (5) 0.0198 (2)
C11 −0.4378 (4) 0.42569 (13) 0.07003 (5) 0.0238 (3)
H11A −0.5449 0.5033 0.0650 0.029*
C12 −0.4494 (4) 0.32378 (14) 0.03538 (5) 0.0262 (3)
H12A −0.5655 0.3214 0.0033 0.031*
C13 −0.2574 (4) 0.23088 (13) 0.05846 (5) 0.0260 (3)
H13B −0.2204 0.1529 0.0441 0.031*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0281 (5) 0.0198 (4) 0.0208 (4) −0.0007 (3) −0.0017 (3) 0.0016 (3)
O2 0.0279 (5) 0.0165 (4) 0.0264 (5) 0.0038 (3) −0.0003 (4) 0.0017 (3)
O3 0.0437 (7) 0.0259 (5) 0.0290 (5) 0.0096 (4) −0.0090 (5) 0.0019 (4)
O4 0.0449 (7) 0.0305 (5) 0.0221 (5) −0.0040 (5) −0.0083 (4) −0.0031 (4)
N1 0.0236 (5) 0.0241 (5) 0.0186 (5) 0.0003 (4) −0.0004 (4) 0.0009 (4)
C1 0.0194 (5) 0.0152 (5) 0.0190 (5) −0.0001 (4) −0.0007 (4) −0.0010 (4)
C2 0.0200 (5) 0.0145 (5) 0.0213 (5) 0.0004 (4) 0.0001 (4) 0.0001 (4)
C3 0.0175 (5) 0.0195 (5) 0.0170 (5) −0.0011 (4) 0.0005 (4) 0.0007 (4)
supporting information
sup-3 Acta Cryst. (2006). E62, o2397–o2398
C5 0.0231 (5) 0.0157 (5) 0.0221 (5) 0.0011 (4) 0.0010 (4) −0.0016 (4)
C6 0.0157 (5) 0.0144 (4) 0.0181 (5) −0.0014 (3) 0.0014 (3) 0.0006 (4)
C7 0.0173 (5) 0.0160 (5) 0.0189 (5) −0.0010 (4) 0.0022 (4) 0.0018 (4)
C8 0.0201 (5) 0.0177 (5) 0.0188 (5) 0.0005 (4) −0.0004 (4) 0.0006 (4)
C9 0.0186 (5) 0.0174 (5) 0.0218 (5) −0.0016 (4) 0.0000 (4) 0.0027 (4)
C10 0.0205 (5) 0.0184 (5) 0.0200 (5) −0.0025 (4) −0.0007 (4) 0.0027 (4)
C11 0.0232 (6) 0.0246 (6) 0.0229 (6) −0.0030 (4) −0.0032 (4) 0.0050 (4)
C12 0.0272 (6) 0.0304 (6) 0.0203 (5) −0.0078 (5) −0.0018 (4) 0.0024 (5)
C13 0.0324 (7) 0.0248 (6) 0.0205 (6) −0.0063 (5) 0.0005 (5) −0.0010 (4)
Geometric parameters (Å, º)
O1—C13 1.3690 (15) C5—C6 1.4005 (16)
O1—C10 1.3736 (15) C5—H5A 0.9300
O2—C7 1.2321 (14) C6—C7 1.4998 (16)
O3—N1 1.2287 (15) C7—C8 1.4702 (16)
O4—N1 1.2274 (14) C8—C9 1.3510 (16)
N1—C3 1.4728 (15) C8—H8A 0.9300
C1—C2 1.3921 (16) C9—C10 1.4262 (17)
C1—C6 1.3960 (16) C9—H9A 0.9300
C1—H1A 0.9300 C10—C11 1.3696 (16)
C2—C3 1.3818 (16) C11—C12 1.422 (2)
C2—H2A 0.9300 C11—H11A 0.9300
C3—C4 1.3882 (17) C12—C13 1.354 (2)
C4—C5 1.3873 (17) C12—H12A 0.9300
C4—H4A 0.9300 C13—H13B 0.9300
C13—O1—C10 106.24 (10) O2—C7—C8 121.18 (10)
O4—N1—O3 123.66 (11) O2—C7—C6 118.58 (10)
O4—N1—C3 118.41 (11) C8—C7—C6 120.24 (10)
O3—N1—C3 117.92 (11) C9—C8—C7 119.21 (11)
C2—C1—C6 120.22 (11) C9—C8—H8A 120.4
C2—C1—H1A 119.9 C7—C8—H8A 120.4
C6—C1—H1A 119.9 C8—C9—C10 127.31 (11)
C3—C2—C1 118.51 (11) C8—C9—H9A 116.3
C3—C2—H2A 120.7 C10—C9—H9A 116.3
C1—C2—H2A 120.7 C11—C10—O1 109.83 (11)
C2—C3—C4 122.86 (11) C11—C10—C9 130.54 (12)
C2—C3—N1 118.34 (11) O1—C10—C9 119.63 (10)
C4—C3—N1 118.77 (10) C10—C11—C12 106.66 (12)
C5—C4—C3 118.00 (11) C10—C11—H11A 126.7
C5—C4—H4A 121.0 C12—C11—H11A 126.7
C3—C4—H4A 121.0 C13—C12—C11 106.27 (12)
C4—C5—C6 120.72 (11) C13—C12—H12A 126.9
C4—C5—H5A 119.6 C11—C12—H12A 126.9
C6—C5—H5A 119.6 C12—C13—O1 111.00 (12)
C1—C6—C5 119.67 (10) C12—C13—H13B 124.5
C5—C6—C7 117.48 (10)
C6—C1—C2—C3 −0.96 (19) C5—C6—C7—O2 −3.30 (17)
C1—C2—C3—C4 0.0 (2) C1—C6—C7—C8 −4.10 (18)
C1—C2—C3—N1 −178.27 (11) C5—C6—C7—C8 176.68 (11)
O4—N1—C3—C2 174.86 (12) O2—C7—C8—C9 −0.86 (19)
O3—N1—C3—C2 −4.38 (18) C6—C7—C8—C9 179.16 (11)
O4—N1—C3—C4 −3.49 (19) C7—C8—C9—C10 178.83 (12)
O3—N1—C3—C4 177.26 (13) C13—O1—C10—C11 0.08 (15)
C2—C3—C4—C5 0.7 (2) C13—O1—C10—C9 −179.42 (12)
N1—C3—C4—C5 178.99 (12) C8—C9—C10—C11 −179.87 (14)
C3—C4—C5—C6 −0.5 (2) C8—C9—C10—O1 −0.5 (2)
C2—C1—C6—C5 1.15 (19) O1—C10—C11—C12 0.03 (16)
C2—C1—C6—C7 −178.05 (12) C9—C10—C11—C12 179.45 (13)
C4—C5—C6—C1 −0.40 (19) C10—C11—C12—C13 −0.12 (16)
C4—C5—C6—C7 178.84 (12) C11—C12—C13—O1 0.17 (17)
C1—C6—C7—O2 175.92 (12) C10—O1—C13—C12 −0.16 (16)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C1—H1A···O2i 0.93 2.48 3.151 (2) 129
C9—H9A···O2 0.93 2.41 2.770 (2) 103
C12—H12A···O3ii 0.93 2.55 3.463 (2) 169