Iododurene
Noudjoud Hamdouni,a* Ouarda Brihi,aMohamed Larbi Medjroubi,aJean Meinnelband Ali Boudjadaa
a
Laboratoire de Cristallographie, De´partement de Physique, Universite´ Mentouri-Constantine, 25000 Mentouri-Constantine, Algeria, andbUMR 6226 CNRS–Universite´ Rennes 1, Sciences Chimiques de Rennes, Equipe Matie`re Condense´e et Syste`mes Electroactifs, 263 Avenue du Ge´ne´ral Leclerc, F-35042 Rennes, France Correspondence e-mail: [email protected]
Received 27 October 2012; accepted 11 November 2012
Key indicators: single-crystal X-ray study;T= 293 K; mean(C–C) = 0.007 A˚; Rfactor = 0.035;wRfactor = 0.039; data-to-parameter ratio = 32.4.
The title compound (systematic name: 1-iodo-2,3,5,6-tetra-methylbenzene), C10H13I, crystallizes in the chiral space group P212121. The I atom is displaced by 0.1003 (5) A˚ from the
mean plane of the ten C atoms [maximum deviation = 0.018 (6) A˚ ]. In the crystal, there are no significant inter-molecular interactions present.
Related literature
For the crystal structure of bromodurene, see: Charbonneauet al. (1964, 1965). For the physical properties of mono-halogenated derivatives of durene, see: Balcou et al.(1965). For standard bond lengths in similar compounds, see: Allen (2002); Hopeet al.(1970).
Experimental
Crystal data
C10H13I
Mr= 260.11
Orthorhombic,P212121
a= 5.5099 (3) A˚
c= 15.1704 (6) A˚
V= 993.34 (8) A˚3
Z= 4
= 3.16 mm
T= 293 K
0.100.050.04 mm
Data collection
Nonius KappaCCD diffractometer Absorption correction: multi-scan
(SADABS; Sheldrick, 1996)
Tmin= 0.140,Tmax= 0.193
26027 measured reflections 2271 independent reflections 2036 reflections withI> 3(I)
Rint= 0.047
Refinement
R[F2> 2(F2)] = 0.035
wR(F2) = 0.039
S= 1.04 3272 reflections 101 parameters 36 restraints
H-atom parameters constrained
max= 0.84 e A˚ 3
min= 0.71 e A˚ 3
Absolute structure: Flack (1983), 925 Friedal pairs
Flack parameter: 0.03 (4)
Data collection: COLLECT (Nonius, 2001); cell refinement:
DIRAX/LSQ(Duisenberget al., 2003); data reduction:EVALCCD
(Duisenberget al., 2003); program(s) used to solve structure:SIR2002
(Burlaet al., 2005); program(s) used to refine structure:CRYSTALS
(Betteridgeet al., 2003); molecular graphics:CAMERON(Watkinet al., 1996) andDIAMOND (Brandenburg, 2012); software used to prepare material for publication:CRYSTALS.
The authors thank the Centre de Diffractome´trie de l’Universite´ de Rennes 1 for the opportunity to collect data on the Nonius Kappa CCD X-ray diffractometer. We would also like to thank Dr Olivier Jeannin for the useful advice he provided.
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: SU2521).
References
Allen, F. H. (2002).Acta Cryst.B58, 380–388.
Balcou, Y., Gre´goire, P. & Meinnel, J. (1965).J. Chim. Phys. PCB,62, 536–538. Betteridge, P. W., Carruthers, J. R., Cooper, R. I., Prout, K. & Watkin, D. J.
(2003).J. Appl. Cryst.36, 1487.
Brandenburg, K. (2012).DIAMOND. Crystal Impact GbR, Bonn, Germany. Burla, M. C., Caliandro, R., Camalli, M., Carrozzini, B., Cascarano, G. L., De Caro, L., Giacovazzo, C., Polidori, G. & Spagna, R. (2005).J. Appl. Cryst.38, 381–388.
Charbonneau, G., Baudour, J., Messager, J. C. & Meinnel, J. (1964).Acta Cryst. 17, 780–781.
Charbonneau, G., Baudour, J., Messager, J. C. & Meinnel, J. (1965).Bull. Soc. Fr. Mine´ral. Cristallogr.88, 147–148.
Duisenberg, A. J. M., Kroon-Batenburg, L. M. J. & Schreurs, A. M. M. (2003).
J. Appl. Cryst.36, 220–229.
Flack, H. D. (1983).Acta Cryst.A39, 876–881.
Hope, H., Knobler, C. & McCullough, J. D. (1970).Acta Cryst.B26, 628–640. Nonius (2001).COLLECT. Nonius BV, Delft, The Netherlands.
Sheldrick, G. M. (1996).SADABS. University of Go¨ttingen, Germany. Watkin, D. J., Prout, C. K. & Pearce, L. J. (1996).CAMERON. Chemical
Crystallography Laboratory, Oxford, England.
Structure Reports Online
supporting information
Acta Cryst. (2012). E68, o3391 [doi:10.1107/S1600536812046557]
Iododurene
Noudjoud Hamdouni, Ouarda Brihi, Mohamed Larbi Medjroubi, Jean Meinnel and Ali
Boudjada
S1. Comment
Substituted benzene compounds are generally liquid at room temperature so relatively few crystal structure analyses of
such compounds have been reported. We have synthesized three mono-halogenated products of duren; chloro-, bromo-
and iododurene. These derivatives are solid at 293 K and their dielectric behavior depends on the nature of the
substituent, and in particular of its mass and volume. Contrary to chlorodurene the title iododurene in the solid phase has
a quite low permeability equal to only 2.6, which changes abruptly at fusion to a value of ca. 4 (Balcou et al., 1965). We
report herein on its crystal structure.
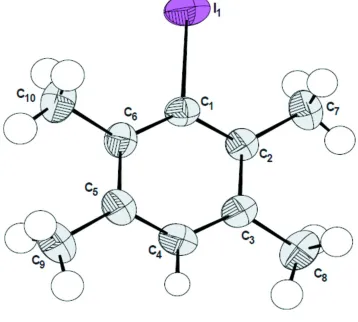
The molecular structure of the title compound is illustrated in Fig. 1. The molecule is almost planar with the I atom
displaced from the mean plane of the nine C atoms [maximum deviation 0.018 (6) Å for the methyl C atoms C7 and C9]
by 0.1003 (5) Å. The average endocyclic angles facing the I atom and the methyl groups are 124.98 (10)° and 117.41
(10)°, respectively. The structural study did not reveal any disorder and the average intramolecular bond lengths, [Car-I =
2.139 (3) Å, Car-Car = 1.400 (5) Å and Car-CMe = 1.498 (6) Å], as well as the average single Car-H bond length [0.940 Å],
agree with the distances reported in the literature [Cambridge Structural Database (Allen, 2002); Hope et al., 1970].
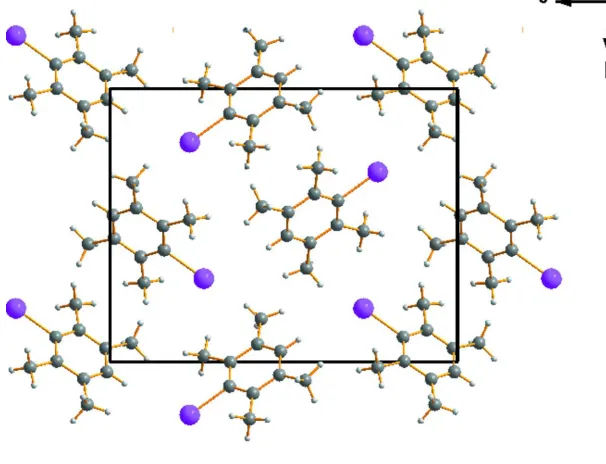
In the crystal, the a axis is very short, so the molecules stack along this direction (Fig. 2). The best mean plane passing
through the carbon and iodine atoms, calculated with the program Molax of CRYSTALS (Betteridge et al., 2003),
indicated that the angle between the normal to this mean plane is ca. 50.1° relative to the a axis, ca. 44.8° relative to the b
axis and ca. 74.5° relative to the c axis (Fig. 3).
The crystals obtained sublimate a little at ambient temperature. Moreover, at room temperature iododurene and the
isotypic product bromodurene (Charbonneau et al., 1964, 1965) crystallize in the same orthorhombic space group with
fixed orientations of the dipolar grouping not presenting dielectric dispersion. This fact could be interpreted by the
importance of the distribution of the masses of the substituents, that inhibit the possibility of molecular reorientation like
in chlorodurene (Balcou et al., 1965).
S2. Experimental
The title compound was synthesized by direct iodination of durene. Equimolecular quantities of iodine and durene were
dissolved in pure acetic acid at 343 K. Progressive iodination was obtained by oxidation of durene using drops of pure
sulfuric and nitric acids diluted in acetic acid. Total precipitation of iododurene was obtained by dilution with water.
Purification was done by chromatography on a silica column and finally precipitation of a solution in chloroform, giving
All H atoms were localized in a difference Fourier map. The C-bound H atoms were included in calculated positions and
treated as riding atoms: C-H = 0.94 Å for CH H atoms, and 0.95 - 0.97 Å for CH3 H atoms, with Uiso(H) = k × Ueq(C)
[image:3.610.126.484.144.466.2]where k = 1.5 for CH3 H atoms and = 1.2 for other H atoms.
Figure 1
The molecular structure of the title molecule, with atom numbering. The displacement ellipsoids are drawn at the 50%
Figure 2
A view along the a axis of the crystal packing of the title compound.
Figure 3
A view along the c axis of the crystal packing of the title compound.
1-Iodo-2,3,5,6-tetramethylbenzene
Crystal data C10H13I
Mr = 260.11
Orthorhombic, P212121
a = 5.5099 (3) Å
b = 11.8839 (5) Å
c = 15.1704 (6) Å
V = 993.34 (8) Å3
[image:4.610.126.429.77.309.2]Dx = 1.739 Mg m−3
Mo Kα radiation, λ = 0.71073 Å
Cell parameters from 8232 reflections
θ = 3.7–27.5°
T = 293 K
Needle, colourless 0.10 × 0.05 × 0.04 mm
Data collection Nonius KappaCCD
diffractometer
Graphite monochromator CCD scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin = 0.140, Tmax = 0.193
26027 measured reflections
2271 independent reflections 2036 reflections with I > 3σ(I)
Rint = 0.047
θmax = 27.5°, θmin = 3.7°
h = −7→7
k = −15→15
l = −19→19
Refinement
Refinement on F
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.035
wR(F2) = 0.039
S = 1.04
3272 reflections 101 parameters 36 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
W = [weight]*[1-(deltaF/6*sigmaF)2]2
(Δ/σ)max < 0.001
Δρmax = 0.84 e Å−3
Δρmin = −0.71 e Å−3
Absolute structure: Flack (1983), 925 Friedal pairs
Absolute structure parameter: −0.03 (4)
Special details
Geometry. Bond distances, angles etc. have been calculated using the rounded fractional coordinates. All su's are estimated from the variances of the (full) variance-covariance matrix. The cell esds are taken into account in the estimation of distances, angles and torsion angles
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
I1 0.93848 (10) 0.80448 (4) 0.27027 (3) 0.0686 (2)
C1 0.9383 (7) 0.9098 (3) 0.1559 (2) 0.0429 (11)
C2 0.7711 (8) 0.9981 (4) 0.1533 (3) 0.0473 (16)
C3 0.7637 (8) 1.0638 (3) 0.0755 (3) 0.0483 (16)
C4 0.9270 (10) 1.0376 (4) 0.0091 (3) 0.0507 (16)
C5 1.0976 (8) 0.9513 (4) 0.0123 (2) 0.0460 (14)
C6 1.1033 (7) 0.8838 (3) 0.0887 (3) 0.0423 (14)
C7 0.5997 (12) 1.0208 (5) 0.2295 (4) 0.071 (2)
C8 0.5866 (12) 1.1574 (5) 0.0651 (4) 0.067 (2)
C9 1.2673 (11) 0.9310 (5) −0.0622 (4) 0.064 (2)
H41 0.92110 1.08070 −0.04290 0.0610*
H71 0.69240 1.03120 0.28260 0.1071*
H72 0.50940 1.08740 0.21780 0.1072*
H73 0.49160 0.95840 0.23690 0.1071*
H81 0.62710 1.21740 0.10510 0.1008*
H82 0.59650 1.18410 0.00600 0.1008*
H83 0.42310 1.13100 0.07630 0.1010*
H91 1.24630 0.98940 −0.10560 0.0971*
H92 1.43080 0.93360 −0.04010 0.0970*
H93 1.23570 0.85840 −0.08810 0.0971*
H101 1.38670 0.80080 0.14830 0.0892*
H102 1.38000 0.78630 0.04650 0.0892*
H103 1.19650 0.71900 0.10410 0.0891*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
I1 0.0831 (3) 0.0727 (3) 0.0500 (2) −0.0082 (2) −0.0036 (2) 0.0120 (2)
C1 0.046 (2) 0.044 (2) 0.0387 (19) −0.006 (2) −0.004 (2) 0.0006 (17)
C2 0.047 (3) 0.044 (2) 0.051 (3) −0.005 (2) 0.000 (2) −0.009 (2)
C3 0.046 (3) 0.040 (2) 0.059 (3) −0.004 (2) −0.011 (2) −0.007 (2)
C4 0.055 (3) 0.045 (2) 0.052 (3) −0.007 (3) −0.011 (3) 0.0031 (19)
C5 0.047 (3) 0.048 (2) 0.043 (2) −0.010 (2) −0.001 (2) −0.0036 (18)
C6 0.041 (3) 0.041 (2) 0.045 (2) −0.0034 (18) −0.0062 (18) −0.0043 (17)
C7 0.070 (4) 0.071 (3) 0.072 (4) 0.000 (3) 0.013 (4) −0.021 (3)
C8 0.063 (4) 0.052 (3) 0.087 (4) 0.008 (3) −0.019 (4) −0.005 (3)
C9 0.060 (4) 0.077 (4) 0.056 (3) −0.009 (3) 0.005 (3) −0.011 (3)
C10 0.056 (3) 0.053 (3) 0.070 (4) 0.011 (3) −0.006 (3) −0.008 (3)
Geometric parameters (Å, º)
I1—C1 2.139 (3) C7—H71 0.9600
C1—C2 1.397 (6) C7—H72 0.9500
C1—C6 1.401 (5) C7—H73 0.9600
C2—C3 1.416 (6) C8—H81 0.9600
C2—C7 1.517 (8) C8—H82 0.9500
C3—C4 1.386 (7) C8—H83 0.9700
C3—C8 1.488 (7) C9—H91 0.9600
C4—C5 1.392 (7) C9—H92 0.9600
C5—C6 1.410 (6) C9—H93 0.9600
C5—C9 1.487 (7) C10—H101 0.9700
C6—C10 1.500 (7) C10—H102 0.9500
C4—H41 0.9400 C10—H103 0.9600
I1—C1—C2 117.6 (3) H71—C7—H72 109.00
I1—C1—C6 117.5 (3) H71—C7—H73 109.00
C2—C1—C6 125.0 (3) H72—C7—H73 110.00
C3—C2—C7 121.3 (4) C3—C8—H83 110.00
C2—C3—C4 117.6 (4) H81—C8—H82 109.00
C2—C3—C8 121.3 (4) H81—C8—H83 110.00
C4—C3—C8 121.1 (4) H82—C8—H83 109.00
C3—C4—C5 125.4 (4) C5—C9—H91 109.00
C4—C5—C6 117.6 (4) C5—C9—H92 109.00
C4—C5—C9 121.2 (4) C5—C9—H93 110.00
C6—C5—C9 121.2 (4) H91—C9—H92 109.00
C1—C6—C5 117.3 (3) H91—C9—H93 110.00
C1—C6—C10 121.5 (4) H92—C9—H93 110.00
C5—C6—C10 121.2 (4) C6—C10—H101 111.00
C3—C4—H41 118.00 C6—C10—H102 109.00
C5—C4—H41 117.00 C6—C10—H103 110.00
C2—C7—H71 109.00 H101—C10—H102 108.00
C2—C7—H72 109.00 H101—C10—H103 110.00
C2—C7—H73 110.00 H102—C10—H103 109.00
I1—C1—C2—C3 −176.8 (3) C7—C2—C3—C4 179.9 (5)
I1—C1—C2—C7 1.5 (6) C7—C2—C3—C8 −0.3 (7)
C6—C1—C2—C3 2.1 (6) C2—C3—C4—C5 0.4 (7)
C6—C1—C2—C7 −179.6 (4) C8—C3—C4—C5 −179.4 (5)
I1—C1—C6—C5 178.1 (3) C3—C4—C5—C6 0.9 (7)
I1—C1—C6—C10 −3.0 (5) C3—C4—C5—C9 −179.2 (5)
C2—C1—C6—C5 −0.8 (6) C4—C5—C6—C1 −0.7 (6)
C2—C1—C6—C10 178.1 (4) C4—C5—C6—C10 −179.6 (4)
C1—C2—C3—C4 −1.8 (6) C9—C5—C6—C1 179.4 (4)