“Typus
Edinburgensis?”
Alexander Habel, M.R.C.P.(UK)
From the Department of Child Life and Health, Edinburgh University
ABSTRACT. An Edinburgh family is reported in which five infants, four female and one male, were found to have a consistently abnormal facial appearance, true or apparent hydrocephalus, retardation in motor and mental develop-ment, failure to thrive and death in the first months of life. Two infants had unexplained neonatal hyperbilirubinemia; the proposita had advanced bone age which may be a pointer to this condition. A superficial similarity to the Brachmann-De Lange syndrome exists, and the important clinical and genetic differences are discussed. Pediatrics, 53:425, 1974,
FAMILIAL RETARDATION-MOTOR AND MENTAL, ABNORMAL
FACIES, HYDROCEPHALUS.
A characteristic facial appearance, failure to thrive, developmental retardation and death within the first year of life occurred in five infants from the same family.
CASE REPORTS
Proposita (IV-4 in Pedigree Fig. 1)
Following a normal pregnancy, an uneventful delivery took place at 38 weeks’ gestation. The infant weighed 4.12 kg and was noted to have abnormal features from birth. J aundice became marked at 48 hours of age. The peak indirect serum bilirubin level was 18.0 mg/100 ml (with a total serum bilirubin level of 18.5 mg/100 ml). Photo-therapy was used and the jaundice settled within three days. There was no evidence of hemlysis, ABO or Rh incom-patibility, of significant infection or enzyme deficiency. From the beginning she was a very poor feeder and unresponsive to handling. Her respirations were noisy and nasal secre-tions copious. On discharge from the maternity hospital at the age of 1 month she weighed 4.36 kg. A month later she was admitted to a hospital weighing 3.36 kg. She had a hoarse cry and noisy breathing. Pallor accentuated a profuse growth of long, silky, black hair (Fig. 2). This extended from the temples to the eyebrows, which were confluent, and to the nape of her neck. Frontal bossing contrasted with the relative underdevelopment of the rest of her face. The anterior fontanelle was 2.5 cm X 1 .5 cm. The occipitofrontal circumference was 39.7 cm, and the crown heel length was 55 cm. Her eyes appeared small. The nose, which deviated to the left, was small with anteverted nostrils; a catheter was passed through each choana without difficulty. The
philtrum appeared slightly longer than usual and there was a “carp mouth,” the lips were thin, there was slight microg-nathia. The trunk and limbs were symmetrically wasted. Fine, dark, downy hair extended down the back. The exter-nal genitalia were normal. Fixed flexion deformities of the elbows were present as well as ulnar deviation of the fingers. The palmar creases were normal. The hands and feet ap-peared relatively long and slender. There was impairment of movement of the right half of the face. Muscle tone was mar-kedly increased, especially in the lower limbs, and she
spon-taneously adopted diplegic postures, with scissoring of the legs and bilateral spontaneous extensor Babinski responses (Fig. 3). The hips were in joint.
She failed to gain weight and to progress beyond the newborn stage of development during her four-week hospital
stay. At 3 months of age she died suddenly during a
pro-longed tonic seizure.
Investigations
Skeletal survey at 3 days of age showed a bone age of three months ( Figs. 4 and 5). At two months of age a lum-bar air encephalogram showed moderate dilatation of the left ventricle and temporal horn. An intravenous pyelogram was normal. All investigations, including chromosome analysis (with and without Giemsa banding), blood and urine amino acid chromatography, were normal: protein bound iodine (PBI, 9.3tg/100 ml; cerebrospinal fluid pro-tein, 16 mg/i#{174} ml; and glucose, 28 mg/100 ml. However, the levels of IgC, 164 mg/100 ml and 234 mg/100 ml (normal range, 272 to 762 mg/100 ml), and IgA, 0 mg/100 ml and 10 mg/100 ml (normal range, 5 to 56 mg/100 ml), were low.
At necropsy, the brain weighed 790 gm (normal for age, 516 gm) and showed microgyria in the frontal and occipital lobes. Microgyria was particularly marked in relation to the posterior ramus of the left Sylvian fissure, which extended posteriorly much further than normal and was unusually deep and wide ( Figs. 8 and 7). The left trigon and occipital horn were slightly enlarged. Microscopic examination also showed occasional small foci of ectopic glia in the lepto-meninges. Particularly noticeable was the widespread paucity
(Received
April 23; revision accepted for publication Octo-ber 21, 1973.)TABLE I
426 TYPUS EDINBURGENSIS
CHARACTERISTIC FEATURES OF THE AFFECTED INFANTS
Feature 11-4 111-4 111-18 IV-2 IV-4 Total
Birth date
Maternal age Sex
Gestational age from last menstrual period (weeks) 10/10/47 22 F 37 17/11/48 23 F 36 3/8/60 23 F 38 1971 23 M 40 28/12/71 22 F 38 Measurements at Birth Weight (kg)
Occipitofrontal circumference (cm) Crown heel length (cm)
2.50 36 49 2.45 36.5 46.5 3.15 36.5 50 3.21 41 50 4.12 36.5 52.5 Physical Characteristics Luxuriant hair Frontal bossing
Functional or relative nasal obstruction
Apparent microphthalmia “Carp mouth” ‘ ‘Low set ears”
Long slender hands, feet and digits
x x x x x x x x x x x x x x x x x x x x x x x x x x x 4/5 5/5 5/5 4/5 4/5 3/5 2/5
Growth Failure to thrive x x x x x 5/5
CNS Profound mental retardation Convulsions
x x
x
x x x
x
5/5 2/5
Jaundice Early, unexplained x x 2/5
Death
Investigations
ge in months
.\rnirio acid chroinatogram (urine) Chromosomes
Wassertnann reaction
4 6 5 8 3
Neg Neg Neg
Normal Normal Neg Normal Normal Neg
of myelin in the centrum semi-ovale of the hemispheres and the cerebral peduncles. Astrocytic nuclei were increased in number. There was no evidence of acute myelin breakdown,
metachromatic bodies, lipid storage or inflammation.
The pharynx, esophagus and trachea contained milk curds,
indicating death had followed aspiration of stomach con-tents. The other internal organs appeared normal.
Infant 2 (lV-2 in Pedigree Fig. 1)
This infant, the only male affected, had long silky auburn
1
FIG. 1. Pedigree of family. Note that 11-5 died in his teens and 111-14 at 11 weeks of age, both suspectedly from
con-genital heart disease. 11-4 died of trauma in his teens.
hair and appeared ‘dysmature’ at birth. Hydrocephalus, on the basis of an O.F.C. of 41 cm at birth, was suspected, and there was frontal bossing, hypertelorism and convergent strabismus. He showed microstomia ( small “triangular” mouth) with micrognathia, brushileld spots, and abnormally differentiated auricles (Figs. 8 and 9). His hands and feet were disproportionately long and slender. Throughout his life he had feeding and respiratory difficulties and was noted to be retarded in development. At 5 months of age an episode of acute congestive cardiac failure occurred. Auscultation of the heart was normal. An x-ray film of the chest showed cardiac enlargement, and an electrocardiograph revealed right ventricular hypertrophy. He died of bronchopneumonia at the age of 8 months weighing only 6 kg, his head cir-cumference having increased to 47 cm. Permission for necropsy was refused.
Infant 3 (llI-3 in Pedigree Fig. 1)
Following cesarian section for prolonged obstructed labor, this female infant displayed cerebral irritability for the
first 24 hours of life. Her respirations were noisy from the start. Weight gain and development were slow. This re-sulted in delaying discharge from the maternity hospital for two months. At the age of 4 months she was admitted to hospital with psychomotor retardation and failure to thrive. She was pale and puny, weighing 3.3 kg. Her hair was like a “syphilitic mop.” She had snuffies. The frontal and parietal bones were prominent and both the anterior and posterior
at Viet Nam:AAP Sponsored on September 8, 2020
www.aappublications.org/news
Fzc. 2. Proposita at two months of age (weight, 3.36 kg). Pallor accentuated a profuse growth of long, silky, black hair.
fontanelles were widely open. The occipitofrontal circum-ference was 44 cm (slightly greater than 98th percentile). Bronchopneumonia was diagnosed and treatment begun, but the infant was removed from hospital, against medical advice, a few days later. She was readmitted within 24 hours with severe dehydration due to vomiting and diarrhea, and she died within the hour.
Necropsy revealed an internal hydrocephalus. The entire ventricular system was dilated from the anterior horns of the lateral ventricles to the fourth ventricle. No adhesions were present and careful inspection of the roof of the fourth ventricle failed to show any lesion. Microscopic examination showed centrilobular fatty degeneration of the liver and congested lungs. Sections of brain tissue were not examined.
No other structural defects or malformations were present.
At necropsy the brain was found to be macroscopically and microscopically normal. Inspection of the internal organs failed to reveal the cause of death.
Infant 5 (lIl-18 in Pedigree Fig. 1)
Two previous pregnancies had been complicated by Rh isoimmunization and was again suspected in the course of mother’s fifth. She was Rh negative, indirect Coombs test positive, with an antiglobulin titer of 1 in 20. Following delivery at 38 weeks’ gestation the infant was found to be Rh negative and direct Coombs test negative. However, the serum bilirubin level rose maximally to 16.5 mg/100 ml on the fourth day of life and settled without treatment. She was discharged home at the age of 2 weeks, weighing 2.9 kg. She required outpatient medical care for persistent severe nasal obstruction and failure to thrive normally. At five months she was admitted with a four-day history of cough and pyrexia. She was found to have hypernatremic dehydration. The BUN was 104 mg/100 ml. Death occurred a few hours later. Necropsy revealed a wasted infant weigh-ing 4.56 kg. Her face was “bird-like,” with frontal bossing
and apparent microphthalmia. The external features of the brain were normal. The cause of death was acute bronchio-litis. The kidneys showed glomerular damage and tubular dilatation, thought to be vaso-occiusive in origin and of long standing.
Genetic Background (Fig. 1)
The pedigree of this Edinburgh family contained no
con-Infant 4 (111-4 in Pedigree Fig. 1)
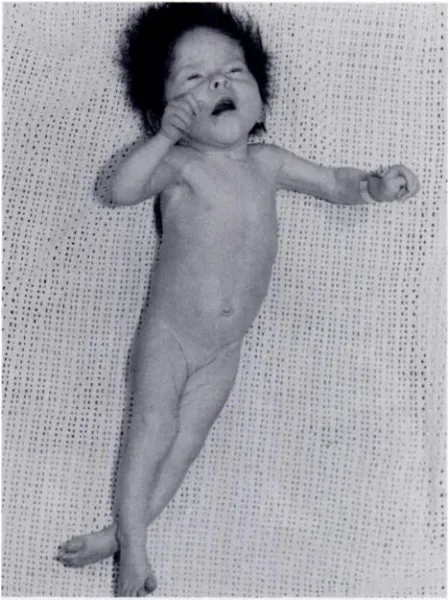
Born a year after her sister, following a difficult delivery, this infant was discharged from the maternity hospital at the age of 6 weeks weighing 2.7 kg. Upper airway obstruc-tion was present from birth. At the age of 5 months a per-sistent erythematous rash developed which covered most of her body. A month later, following a series of convulsions, she was admitted to hospital. The striking resemblance to her sister was noted. Her occipitofrontal circumference was 42 cm; weight, 4 kg; length, 56 cm (Fig. 10). She was treated for suspected bronchopneumonia. The cerebrospinal
fluid pressure was greater than 300 cm of water, and the constituents were normal. An x-ray film of the skull re-vealed no evidence of raised intracranial pressure. A week after admission an air encephalogram suggested a mild
internal hydrocephalus. She died suddenly three days later.
Fic. 3 Photograph of proposita. Muscle tone was markedly increased, especially in the lower limbs, and she spontan-eously adopted diplegic postures, with scissoring of the legs
428 TYPUS EDINBURGENSIS
FIG. 4. Proposita: skeletal survey at 3 days of age showed
a bone age of three months.
sanguineous marriages. Congenital heart disease may have occurred in two members of the family ( I, 5:11, 16 ) but hospital notes were no longer available to confirm this. One
male in generation I died from trauma (I, 4)
.
Careful exam-ination of the parents of the proposita failed to reveal anyIlmnifestations of the syndrome reported.
FIG. 6. At necropsv, the brain showed microgvria which was particularly marked in relation to the posterior ramus of the left Sylvian fissure, which extended posteriorly much further
than normal and was unusually deep and wide.
Fic. 5. Proposita: skeletal survey at 3 days of age showed
a bone age of three months.
DISCUSSION
The facial features of the infants reported have
a certain similarity to the Brackmann-De Lange
syndrome. The latter have a readily recognizable facial appearance from birth, suffer from intra-uterine growth retardation and fail to thrive. They are often microbrachycephalic with small, poorly developed brains, and suffer severe mental retarda-tion. Reduction deformities of the limbs are com-mon. Highly significant findings include minor
abnormalities of the fifth finger, the presence of single palmar creases, “proximally placed” thumbs,
microcephaly, abnormal voice, mouth and lips, and a family history of a similarly affected sibling.1 Bone age is usually retarded. Hypoplasia of the
genitals, cardiac defects and dwarfing of the in-ternal organs are not uncommonly found at necropsy. Survival beyond the first year of life is usual.2 In contrast, all five infants in the family reported were of at least average height and weight for gestational age and their head circumferences were on or above the mean at birth
(
Table I)
. Thehands and feet were long and slim with no
abnor-malities of the fifth finger or palmar flexion crease.
at Viet Nam:AAP Sponsored on September 8, 2020
www.aappublications.org/news
Fic. 7. Close-up of Figure 6.
All the infants died before attaining the age of 1 year and at necropsy two had abnormally large brains’ : one was hydrocephalic, the other showed micropolygyria. No structural congenital
abnormali-ties of other organs, or hypoplasia of the genitals, was found. The unusually advanced osseous matura-tion of the proposita is the reverse of that observed
in the BDLS. Uncommon in the neonatal period, it has been described in hyperthyroidism,
congeni-Fic. 9. Microstomia with micrognathia, brushfield spots and abnormally differentiated auricles.
tal adrenal hyperplasia, peripheral dysostosis and a syndrome affecting unrelated males who have char-acteristic facies with failure to thrive.4 Advanced
bone age may, therefore, be a marker to the conch-tion described.
The five infants’ appearance was strikingly simi-lar, resembling each other more closely than their
unaffected siblings. It can be concluded that they all had the same underlying pathology. Whether this was due to a primary central nervous system
disorder, or a collection of apparently unrelated multiple congenital anomalies forming a syndrome
complex, cannot be determined on the present clinical evidence.
The mode of inheritance of the condition de-scribed is conjectural. A simple Mendelian
domi-nant mutation seems precluded in view of the early death of the affected infants. An autosomal reces-sive trait, as suggested for inheritance of the BDLS by Opitz and Grosse,5 would require that an
ab-normal gene was not only fairly common in the
general population but that four separate
heterozy-Fic. 8. Microstomia ( small “triangular” mouth ) in infant 2. gotes should marry into the same family, an
at Viet Nam:AAP Sponsored on September 8, 2020
www.aappublications.org/news
430 TYPUS EDINBURGENSIS
number of affected infants and those most likely
to propagate the trait. As three of 18 individuals
were abnormal the risk of recurrence in affected families is 17%. If the transmission is through a chromosome abnormality as discussed above, then the risk is halved for the siblings of these infants and the likelihood of recurrence is then 8 to 9%.
The eponym “Typus Amstelodamensis” was
coined by Cornelia de Lange to describe the BDLS.
She derived it from the name of the city in which the children lived. The facial similarity between
those children and the infants born into an Edin-burgh family is remarkable, yet the differences in inheritance of the trait, its physical characteristics and clinical course sufficiently distinct, that it should be considered a separate entity and there-fore “Typus Edinburgensis.”
ADDENDUM
The mother of the proposita has had two further pregnancies, both resulting in spontaneous abortion at three months’ gestation. The aborted material was not available for examination.
FIG. 10. Face in infant 4. Note striking resemblance to her sister.
likely occurrence as this is the first family known to be affected with this syndrome. The absence of
a “forme-fruste” and the presence of skipped
gen-erations in branches of the family tends to
ex-dude an autosomal dominant gene with variable expressivity.
The observed frequency of occurrence of the
syndrome
(
Fig. 1)
is best explained by theinheri-tance of a chromosome abnormality submicroscopic
in type and therefore undetected by present day
methods of analysis. This abnormality, in the form
of a small insertion or translocation, may be present
on heterologous chromosomes in healthy carriers
leading to unbalanced homologous pairs in the
off-spring. Half the gametes of such a parent would be
expected to confer carrier status or give rise to an affected infant. In the absence of such a demonstra-hle chromosomal abnormality it is only possible
to compute an empirical risk. Generation III was chosen for this purpose. It contains the largest
REFERENCES
1. Motl, NI. L., and Opitz, J. I. : Studies of malformation
syndromes XXVA. Hum. Hered., 21:1, 1971. 2. Berg, J. Nt, McCreary, B. D., Ridler, MAC., and Smith,
C. F. : The De Lange Syndrome. Institute for Research into Mental Retardation Monograph No. 2. Oxford: Pergamon Press Ltd., 1970.
3. Dekaban, A. : Neurology of Infancy. Baltimore: Williams and Wilkins Co., 1959, pp. 8. 21.
4. Marshall, R. C., Crahani, C. B., Scott, C. R., and Smith, D. W. : Syndrome of accelerated skeletal maturation and relative failure to thrive: A newly recognized clinical growth disorder. J. Pediat., 95:95, 1971. 5. Opitz, J. M., and Crosse, F. R. : Year Book of Pediatrics,
1971. Chicago: Year Book Medical Publishers Inc., 1971, pp. 486-491.
ACKNOWLEDGMENT
I wish to thank Dr. A. F. j. Maloney and Dr. A. Cordon of the Pathology Department, Royal Infirmary, Edinburgh,
for performing the neuropathological examination of the proposita; Dr. W. J. Matheson, Leicester Royal Infirmary, for the photographs and clinical details of IV-2; and Dr. D. vl. Douglas, Pediatrician, and Dr. A. D. Bain, Pathologist, of the Royal Hospital for Sick Children for permission to
report this family. I am particularly indebted to Dr. Matilda Nelson, formerly of the University Department of Human Genetics, Edinburgh, who gave genetic advice and read the
draft of this article.
at Viet Nam:AAP Sponsored on September 8, 2020
www.aappublications.org/news
1974;53;425
Pediatrics
Alexander Habel
"Typus Edinburgensis?"
Services
Updated Information &
http://pediatrics.aappublications.org/content/53/3/425
including high resolution figures, can be found at:
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtml
entirety can be found online at:
Information about reproducing this article in parts (figures, tables) or in its
Reprints
http://www.aappublications.org/site/misc/reprints.xhtml
1974;53;425
Pediatrics
Alexander Habel
"Typus Edinburgensis?"
http://pediatrics.aappublications.org/content/53/3/425
the World Wide Web at:
The online version of this article, along with updated information and services, is located on
American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.
American Academy of Pediatrics, 345 Park Avenue, Itasca, Illinois, 60143. Copyright © 1974 by the
been published continuously since 1948. Pediatrics is owned, published, and trademarked by the
Pediatrics is the official journal of the American Academy of Pediatrics. A monthly publication, it has
at Viet Nam:AAP Sponsored on September 8, 2020
www.aappublications.org/news