Divya Yada et al. J Sci Res Pharm, 2017;6(10):117-124
World Inventia Publishers
J
ournal of
S
cientific
R
esearch in
P
harmacy
http://www.jsrponline.com/
Vol. 6, Issue 10, 2017 ISSN: 2277-9469
USA CODEN: JSRPCJReview Article
IMPURITY PROFILING: AN EMERGING TASK TO PHARMACEUTICAL INDUSTRIES NOW A DAYS
Divya Yada *, T. Rajeshwari, Dr. M. SudhakarMallareddy College of Pharmacy, Maisammaguda, Secunderabad-500014, Telangana, INDIA.
Received on: 02-10-2017; Revised and Accepted on: 23-10-2017
ABSTRACT
I
mpurity is defined as any foreign substance coexisting with the original drug, it may arises from starting material or intermediates i.e., due to any side reactions. Characterization and control of impurities in pharmaceutical substances and products are reviewed with significant issues specific to the active pharmaceutical ingredient and pharmaceutical formulations. The control of pharmaceutical impurities in the pharmaceutical industry is crucial task to the formulator. The International Conference on Harmonization (ICH) has formulated a workable guideline regarding the control of impurities in pharmaceutical drug substance as well as formulations. In this review article, impurities, its different types and origin of impurities, qualification of impurities and analytical method development have been discussed briefly.KEYWORDS: Impurity Profiling, Emerging Task, Pharmaceutical Industries.
INTRODUCTION
I
mpurity is defined as any foreign substance coexisting with the original drug, it may arises from starting material or intermediates i.e., due to any side reactions. A substance is said to be impure, one that affects the purity of the material of interest, viz., an active pharmaceutical ingredient (API) or drug substance. The primary goal of drug development is to control Impurity levels in pharmaceuticals. The purity of a drug product is in turn determined on the basis of the percentage of the labeled amount of API found in it by a suitable analytical method.In general, impurities are small molecules and are especially found in solid dosage forms where the limited mobility restricts the reactivity of larger molecules. In most of the drugs, the reactive species consist of water (which can hydrolyze or effect the performance of dosage form), small electrophiles (e.g., aldehyde and carboxylic acid derivatives), peroxides (oxidizes some drugs), and metals (that catalyses oxidation and other drug degradation pathways). Apart from these effects, some impurities can cause toxicological problems. The presence of these unwanted chemicals, even in small amounts, may influence the efficacy and safety of API. Degradation product impurities along with process-related impurities are necessary to control in APIs. Potential impurities arises during API manufacturing process includes starting materials, isomers, intermediates, reagents, solvents, catalysts and reaction byproducts.
The main objective of investigating potential impurities is to determine process control mechanisms for their removal and need for specification controls at appropriate points in the process. Several descriptions are enclosed in the literature for impurity investigations and method development to support them [1, 2]. By means of impurity
profile, analytical activities of detecting, identifying or elucidating the structure and quantitatively determining organic and inorganic impurities as well as residual solvents in bulk drugs and pharmaceutical formulations [2].
*Corresponding author:
Divya Yada
Mallareddy College of Pharmacy,
Maisammaguda, Secunderabad-500014, Telangana, INDIA.
* E-Mail: [email protected]
Quantitative determination of these impurities served as a method for the quality control and validation of drug substances. Regulatory authorities such as US FDA (United States Food and Drug Administration), CGMP (Current Good Manufacturing Practice), TGA (Thermo Gravimetric Analysis), and MCA (Ministry of Corporate Affairs) insist on the impurity profiling of drugs.
Impurities in new drug substances can be addressed from two aspects:-
(1) The chemical aspect, which includes classification and identification of impurities, report generation, listing of impurities in specifications, and a brief discussion of analytical procedures.
(2) The safety aspect, which includes specific guidance for quantifying impurities, present, substantially at lower levels, in a drug substance used in clinical studies [3].
Impurity profile and drug safety:
The safety of drug therapy is related to the quality of drugs. The safety requirements with respect to API content of bulk-drug materials in various pharmacopoeias are usually in the range of 98–99%
[4-6]. The main aim is to minimize the adverse effects (AE) of drug
materials and the other preparations made from it.
Impurity profile is description of the identified and unidentified impurities present in a typical batch of API produced by a specific controlled production process8-10. It is one of the most important fields of activity in contemporary industrial pharmaceutical analysis. The main reasons for the increasing interest of drug manufacturers and drug registration authorities in the impurity profiles of bulk drug substances are as follows: [7]
a) In the course of the development of a new drug or a new technology for manufacturing an existing drug it is essential to know the structures of the impurities: by possessing the information synthetic organic chemists are often able to change the reaction conditions in such a way that the formation of the impurity can be avoided or its quantity reduced to an acceptable level.
b) Having suggested structures for the impurities, they can be synthesized and thus provide final evidence for their structures previously determined by spectroscopic methods.
determination of the impurity and the use of this method as part of the quality control testing of every batch.
d) In case of major impurities the synthesized or isolated material can be subjected to toxicological studies thus greatly contributing to the safety of drug therapy.
e) For drug authorities the impurity profile of a drug substance is a good fingerprint to indicate the level and constancy of the manufacturing process of the bulk drug substance.
After establishing the pharmacological toxicological profile of a drug substance, pharmacologists, clinicians and drug-registration authorities consider its beneficial and adverse effects to the human organism and, on the basis of the benefit/risk ratio thus obtained, make the decision with respect to the possibility of introducing it into therapy.
Common terms for impurities: [8]
1. Intermediate, Penultimate intermediate and Byproducts 2. Transformation products
3. Interaction product 4. Related product 5. Degradation product
1. Intermediate, Penultimate Intermediate and Byproducts: The compounds produced during synthesis of the desired material are called intermediates, especially if they have been isolated and characterized. The penultimate intermediates are the last compound in the synthesis chain prior to the production of the final
desired compound. Byproducts are unplanned compounds produced in between the reaction. It may or may not be possible to theorize all of them.
2. Transformation Products:
They are very similar to by-products which relates to theorized and non-theorized products that may be produced in the reaction.
3. Interaction products:
Interaction products that could occur between various involved chemicals intentionally or unintentionally.
4. Related Products:
These products have similar chemical structure and potentially similar biological activity.
5. Degradation products:
These compounds are products due to decomposition of the active ingredient or the material of interest.
According to ICH guidelines, impurities in drug substance produced by chemical synthesis can be broadly classified into following three categories
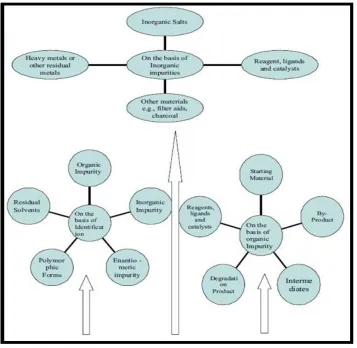
1.Organic Impurities (Process and drug-related) 2.Inorganic Impurities (Reagent, ligands, catalysts) 3.Residual Solvents (Volatile solvents)
Fig. 1: Sources of Impurities
Organic Impurities:
Organic impurities may arise during the manufacturing process and/or storage of the drug substance. They may be identified or unidentified, volatile or non-volatile, and these include the starting material, intermediates, degradation products, by-products and reagents, ligands and catalyst used at different stages of synthesis of API and drug products. These include following sub-impurities.
Starting Materials or Intermediate Impurities:
By-products:
In synthetic organic chemistry, getting a single end product with complete yield is very rare; there is always a chance of having by products along with desired end product.
Degradation Products:
Impurities can also be formed by degradation of the end product during manufacturing of bulk drugs. This mainly occurs due to improper storage of formulation.
Chemical reagents, ligands and catalysts:
Chemical reagents, ligands and catalysts used in the synthesis of a drug substance can be carried over to the final products as trace level impurities. For e.g. carbonic acid chloromethyl tetrahydro-pyran-4-yl ester (CCMTHP), which is used as an alkylating agent in the synthesis of a β-lactam drug substance, was observed in the final product as an impurity. Many chemical reactions are promoted by metal based catalysts. For instance, a Ziegler-Natta catalyst contains titanium, Grubb’s catalyst contains ruthenium and Adam’s catalyst contains platinum. In some cases, reagents or catalysts may react with intermediates or final products to form byproducts. For e.g., Pyridine, a catalyst used in the course of synthesis of mazipredone, reacts with an intermediate to form a pyridinium impurity [9].
Other Types of Organic Impurities: [10-17]
A. Synthesis Related Impurities:
New chemical entity generated during synthetic process from raw material, solvent, intermediate, byproduct. During synthesis process, if impurity present in trace or in significant amount in any of substance involved in reaction, that ultimately result in final product contaminated with one or more unwanted materials. Therefore, synthesis related impurity require upmost care during every step involved in synthesis process to minimize level of impurity that can arise.
B. Formulation Related Impurities:
Drug substance subjected to variety of conditions that leads to its degradation or other reactions. Solutions and suspensions are prone to degradation due to hydrolysis. Water used in formulation contribute to not only its impurity but also provide situation for hydrolysis and catalysis.
Factors Affecting On Formulation Related Impurities:
a. Environment related:
I. Exposed to adverse temperature: Substance which are labile to heat or in tropical temperature lead to degradation of active constitute and formation of impurity occurs. E.g. Vitamins are heat sensitive and its degradation lead to loss in potency. II. Exposed to light: Photosensitive material when exposed to light
/ UV light undergo degradation which forms impurity.
III. Humidity: It can be detrimental to bulk powder and formulation containing solid dosage form.
b. Formation of impurities on ageing:
Mutual interaction: Interaction between ingredients involved in formulation leads to mutual interaction which causes impurity formation.
C. Functional Group Related Impurities:
a) Ester hydrolysis: Drugs like aspirin, benzocaine, cefoxime, cocaine, ethyl paraben undergo ester hydrolysis.
b) Hydrolysis: Commonly drugs like benzyl penicillin, barbital, and chloramphenicol undergo hydrolysis.
c) Oxidative degradation: Drugs like hydrocortisone, methotrexate, heterocyclic aromatic ring, nitroso/nitrile derivative.
d) Photolytic cleavage: Product exposed to light while manufacturing or storage in hospital pending use or by consumer pending use.
e) Decarboxylation: Some dissolved carboxylic acid such as p-amino salicylic acid loose CO2 when heated
Inorganic Impurities:
Inorganic impurities can result from the manufacturing process. They are normally known and identified.
a. Reagent, Ligands and Catalysts: The chances of having these impurities are rare however, in some processes; these could create a problem unless the manufacturers take proper care during production.
b. Heavy Metals:Water is generally used in different manufacturing processes which act as the main source of heavy metals, like Ar, Cd, Cr, Na, Mg, Mn, etc (if stainless steel reactors are used) where acidification or acid hydrolysis takes place. By using demineralized water and glass-lined reactors heavy metal impurities can be easily avoided.
c. Other Materials (Filter Aids, Charcoal):The filters or filtering aids such as centrifuge bags are routinely used in the bulk drugs manufacturing plants and in many cases, activated carbon is also used which also act as a source of impurity. Therefore regular monitoring of fibers and black particles in the bulk drugs is essential to avoid contamination.
Residual Solvents:
Residual solvents are organic volatile chemicals that are used or produced in the manufacture of drug substances or excipients, or in the preparation of drug products. Residual solvents are organic or inorganic liquids used during the manufacturing process. It is very difficult to remove these solvents completely by the work-up process. Some solvent that are known to cause toxicity should be avoided in the production of bulk drugs.
The residual solvents [18] are classified as follows:
Class 1 solvents:Solvents to be avoided in pharmaceutical products Known human carcinogens, strongly suspected human carcinogens and environmental hazards. Example was shown in Table 1
Class 2 solvents: Solvents to be limited in pharmaceutical products:
Non-genotoxic animal carcinogens or possible causative agents of other irreversible toxicity such as neurotoxicity or teratogenicity. Solvents suspected of other significant but reversible toxicities. Example was shown in Table 2
Class 3 solvents: Solvents with low toxic potential:Solvents with low toxic potential to man; no health -based exposure limit is needed. These solvents are less toxic in acute or short term studies and negative in genotoxic studies. The amount of these residual solvents of 50mg or less would be acceptable. Examples: Acetic acid, Acetone, Anisole, 1-Butanol, 2-Butanol etc.
Class 4 solvents: Solvents for which No adequate toxicological data was found: The solvents may be of interesting to manufacturers of excipients, drug substances or drug products. But there was no adequate toxicological data on which to base a Permitted Daily Exposure was found. Examples: 1,1-Diethoxy propane,1,1-Dimethoxy propane, 2,2-Dimethoxy propane, Isooctane etc.
Table No. 1: Class 1 solvents in pharmaceutical products (solvents that should be avoided)
Solvent Concentration limit (ppm) Concern
Benzene 2 Carcinogenic
Carbon Tetra chloride 4 Toxic and environmental Hazard
1,2-Dichloroethane 5 Toxic
1,1-Dichloroethane 8 Toxic
Table No. 2: Class 2 solvents in pharmaceutical products (solvents that should be avoided)
Solvent Concentration limit (ppm) PDE (mg/day)
Acetonitrile 410 4.1
Chlorobenzene 360 3.6
Chloroform 60 0.6
Cumene 70 0.7
Cyclohexane 3880 38.80
Dichloromethane 1870 18.70
Dioxane 380 3.8
Other Impurities:
Enantiomeric impurities: The single enantiomeric form of a chiral drug is now considered as an improved chemical entity that may offer a better pharmacological profile and an increased therapeutic index, with a more favorable adverse reaction profile. However, the pharmacokinetic profiles of levofloxacin (S-isomeric form) and ofloxacin (R-isomeric form) are comparable, suggesting the lack of advantages of a single isomer in this regard. For the manufacturers of a single enantiomeric drug (eutomer), the undesirable stereoisomers in drug control are considered in the same manner as other organic impurities
[19].
Crystallization related impurities: Impurity can be any substance other than the material being crystallized. Therefore, even the solvent from which the crystals are grown can be considered as an impurity. When impurities are added specifically to produce a desired morphological effect they are referred to as additives. The presence of impurities or additives in a crystallization system can have a radical effect on crystal growth, nucleation, and agglomeration, as also on the uptake of foreign ions in the crystal structure [20].
Stereochemistry related impurities:It is of paramount importance to look for stereochemistry related compounds; that is, those compounds that have a similar chemical structure, but different spatial orientation. These compounds can be considered as impurities in the APIs. The single enantiomeric form of a chiral drug is now considered as an improved chemical entity that may offer a better pharmacological profile and an increased therapeutic index, with a more favorable adverse reaction profile, for example, the pharmacokinetic profile of levofloxacin (S-isomeric form) and ofloxacin (R-isomeric form) are comparable, other examples are levofloxacin (S-ofloxacin), esomeprazole (S-omeprazole), and lavalbuterol (R-albuterol) [19].
Formulation related impurities (impurities in drug products):
Number of impurities in a drug product can arise out of inert ingredients used to formulate a drug substance. In the process of formulation, a drug substance is subjected to a variety of conditions that can lead to its degradation or other deleterious reaction. Solutions and suspensions are potentially prone to degradation due to hydrolysis. Water used in the formulation cannot only contribute its own impurities; it can also provide a ripe situation for hydrolysis and catalysis. Similar reactions are possible in other solvents that may be used.
The formulation related impurities can be classified as follows:
1) Method related 2) Environmental related
The primary environmental factors that can reduce stability include the following
I. Exposures to adverse temperatures II. Light-especially UV light
III. Humidity
Formation of impurities on ageing
I. Mutual interaction amongst ingredients II. Functional group- related typical degradation
i) Ester hydrolysis ii) Hydrolysis
iii) Oxidative degradation iv) Photolytic cleavage v) Decarboxylation
Qualification of Impurities: [21]
Qualification is the process of acquiring and evaluating data that establishes the biological safety of an individual impurity or a given impurity profile at the level(s) being considered. When appropriate, we recommend that applicants provide a rationale for establishing impurity acceptance criteria that includes safety considerations.
An impurity is considered qualified when it meets one or more of the following conditions:
• When the observed level and proposed acceptance criterion for the impurity do not exceed the level observed in an FDA approved human drug product.
• When the impurity is a significant metabolite of the drug substance.
• When the observed level and the proposed acceptance criterion for the impurity are adequately justified by the scientific literature. • When the observed level and proposed acceptance criterion for the
impurity do not exceed the level that has been adequately evaluated in comparative in vitro genotoxicity studies.
The International Conference on Harmonization (ICH) addresses questions relating to impurities as follows:
• Q1A (R) stability testing of new drug substances and products. • Q3A (R) impurities in drug substances.
• Q3B (R) impurities in drug products. • Q3C impurities: residual solvents.
• Q6A specifications: test procedures and acceptance criteria for new drug substances and new drug products; chemical substances.
Limits for impurities:According to ICH guidelines on impurities in new drug products, identification of impurities below 0.1% level, is not considered to be necessary, unless potential impurities are expected to be unusually potent or toxic. Recommended qualification thresholds based on the maximum daily dose as described in Table No. 3 for drug substance and Table No. 4 for drug product, are provided in ICH Q3A [22]
and ICH Q3B [23].
Table No. 3: Drug substance impurities thresholds
Maximum daily dosea+ Reporting thresholdb,c Identification thresholdb,c Qualification thresholdb,c
≤ 2g/day 0.05% 0.10% or 1.0 mg/day intake
(whichever is less) 0.15% or 1.0 mg/day intake (whichever is less)
≥ 2g/day 0.03% 0.05% 0.05%
Table No. 4: Thresholds for degradation products in drug products
Maximum daily dosea Reporting thresholdb,c
≤1 g 0.1%
>1 g 0.05%
Maximum daily dosea Identification thresholdb,c
<1 mg 1.0% or 5 µg TDI, whichever is lower 1 mg–10 mg 0.5% or 20 µg TDI, whichever is lower
>10 mg–2 g 0.2% or 2 mg TDI, whichever is lower
>2 g 0.10%
Maximum daily dosea Qualification thresholdb,c
<10 mg 1.0% or 50 µg TDI, whichever is lower 10 mg–100 mg 0.5% or 200 µg TDI, whichever is lower
>100 mg–2 g 0. 2% or 3 mg TDI, whichever is lower
>2 g 0.15%
a - The amount of drug substance administered per day; b - Thresholds for degradation products are expressed either as a percentage of the drug substance or as total daily intake (TDI) of the degradation product. Lower thresholds can be appropriate if the degradation product is unusually toxic; c - Higher thresholds should be scientifically justified
Acceptance criteria for impurities:
For newly synthesized drug substances, the specification should include acceptance criteria for impurities. Stability studies, chemical development studies, and routine batch analyses can be used to predict those impurities likely to occur in the commercial product. A rationale for the inclusion or exclusion of impurities in the specification should include a discussion of the impurity profiles observed in batches under consideration, together with a consideration of the impurity profile of material manufactured by the proposed commercial process. For impurities known to be unusually potent or to produce toxic or unexpected pharmacological effects, the quantization or detection limit of the analytical methods should commensurate with the level at which the impurities need to be controlled. Appropriate qualitative analytical descriptive label included in the specification of unidentified impurities. A general acceptance criterion of not more than 0.1 % for any unspecified impurity should be included. Acceptance criteria should be
set, based on data generated on actual batches of the drug substance, allowing sufficient latitude to deal with normal manufacturing and analytical variation, and the stability characteristics of the drug substance. Although normal manufacturing variations are expected, significant variation in batch to- batch impurity levels could indicate that the manufacturing process of the drug substance is not adequately controlled and validated. The acceptance criteria should include limits for organic impurities; each specified identified impurity, each specified unidentified impurity at or above 0.1%, and any unspecified impurity, with a limit of not more than 0.1%, total impurities, residual solvents and inorganic impurities. If data are not available to qualify the proposed specification level of an impurity, studies to obtain such data may be needed (when the usual qualification threshold limits given below are exceeded). According to ICH, the maximum daily dose qualification threshold is considered as follows: ≤ 2g/day 0.1 % or 1 mg per day intake (whichever is lower) ≥2g/day 0.05%.
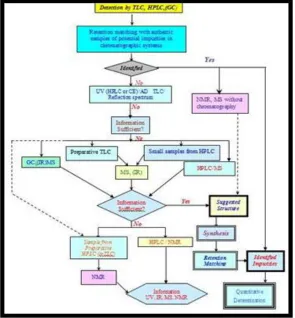
Fig. 2: schematic representation of scheme for impurity profiling of drugs
In the case of unsuccessful identification with standard samples the most reasonable way to determine the structure of impurity starts with the investigation of the UV spectra, easily obtainable with the aid of the diode array detector in the case of HPLC and the quantification with the help of densitometer. In exceptional cases, with full knowledge of the synthesis of drug martial, the structure of the impurity can be
derivatization reaction can be confused with the impurities. The next step in the impurity profiling is the synthesis of the material (impurity standard) with the proposed structure. The retention and spectral matching of the synthesized material with the impurity in question is carried out as outlined above.21 the possibilities of spectroscopic
techniques in drug impurity profiling without chromatographic separation are also worth mentioning. Spectra obtained by using high-resolution, highly sensitive NMR spectrometers and mass spectrometers with APCI /ESI ( Electromagnetic Source Imaging) facilities are suitable to provide a fingerprint picture regarding the purity of the sample.
Fig. 3: General Scheme for Impurity Profiling
Analytical Method Development:
New drug development requires meaningful and reliable analytical data to be produced at various stages of the development.
a) Sample set selection for analytical method development
b) Screening of Chromatographic conditions and Phases, typically using the linear solvent- strength model of gradient elution c) Optimization of the method to fine-tune parameters related to
ruggedness and robustness. The impurities can be identified predominantly by following methods;
• Reference standard method • Spectroscopic method • Separation method • Isolation method • Characterization method
Reference standard method:
The key objective of this is to provide clarity to the overall life cycle, qualification and governance of reference standards used in development and control of new drugs. Reference standards serve as the basis of evaluation of both process and product performance and are the benchmarks for assessment of drug safety for patient consumption. These standards are needed, not only for the active ingredients in dosage forms but also for impurities, degradation products, starting materials, process intermediates, and excipients.
Spectroscopic methods:
The UV, IR, MS, NMR and Raman spectroscopic methods are routinely being used for characterizing impurities.
Separation methods:
The Capillary electrophoresis (CE), Chiral Separations, Gas Chromatography (GC), Supercritical Fluid Chromatography (SFC), TLC, HPTLC, HPLC are regularly being used for separation of impurities and degradation products.
Isolation methods:
It is often necessary to isolate impurities. But if the instrumental methods are used, isolation of impurities is avoided as it directly characterizes the impurities. Number of methods can be used for isolation and characterization of impurities. But the application of any method depends on the nature of impurity (i.e.) its structure, physicochemical properties and availability. Isolation should be initiated based on simple extraction or partition methods. It may be possible to extract impurities selectively on the basis of acidity, basicity, or neutrality.
The following methods are commonly used for the isolation, they are
1. Extraction
2. Column Chromatography 3. Preparative Separations
Extraction:
The extraction process usually involves liquid–liquid extraction, where one phase is an aqueous solution and the other is an organic phase that is nonpolar.
1. Liquid-Solid extraction 2. Liquid-Liquid extraction
The term ‘chromatographic reactor’ refers to the use of an analytical-scale column as both a flow through reactor, and simultaneously, as separation medium for the reactant(s) and product(s).
List of methods that can be used for isolation of impurities are: • Solid-phase extraction methods
• Liquid-liquid extraction methods • Accelerated solvent extraction methods • Supercritical fluid extraction
• Column chromatography • Flash chromatography • TLC
• GC • HPLC • HPTLC
• Capillary electrophoresis (CE)
• Supercritical fluid chromatography (SFC)
Characterization methods:
Highly sophisticated instrumentation, such as MS attached to a GC or HPLC, are inevitable tools in the identification of minor components (drugs, impurities, degradation products, metabolites) in various matrices. For characterization of impurities, different techniques are used; which are as follows;
NMR:
The ability of NMR to provide information regarding the specific bonding structure and stereochemistry of molecules of pharmaceutical interest has made it a powerful analytical instrument for structural elucidation. The ability of NMR- based diffusion coefficient determination to distinguish between monomeric and dimeric substances was validated using a standard mixture of authentic materials containing both monomers and dimers [39]. Unfortunately, NMR has traditionally been used as a less sensitive method compared to other analytical techniques. Conventional sample requirements for NMR are on the order of 10 mg, as compared with MS, which requires less than 1 mg.
MS:
It has an increasingly significant impact on the pharmaceutical development process over the past several decades. Advances in the design and efficiency of the interfaces, that directly connect separation techniques with Mass Spectrometers have afforded new opportunities for monitoring, characterizing, and quantification of drug related substances in active pharmaceutical ingredients and pharmaceutical formulations. If single method fails to provide the necessary selectivity, orthogonal coupling of chromatographic techniques such as HPLC-TLC and HPLC-CE, or coupling of chromatographic separations with information rich spectroscopic methods such as HPLC-MS or HPLC-NMR may need to be contemplated, but hopefully only as a development tool rather than a tool for routine QC use.
Hyphenated Methods: • LC-MS-MS • HPLC-DAD-MS • HPLC-DAD-NMR-MS • GC-MS
• LC-MS
A common goal for investigation of both process and product degradation-related impurities is to determine which of the many potential impurities are, in fact, produced in the manufacturing process and which occur under a given set of storage conditions.
Applications:
Numerous applications have been sought in the areas of drug designing and in monitoring quality, stability, and safety of pharmaceutical compounds, whether produced synthetically, extracted from natural products or produced by recombinant methods. The applications include alkaloids, amines, amino acids, analgesics, antibacterials, anticonvulsants, antidepressant, tranquilizers, antineoplastic agents, local anesthetics, macromolecules, steroids, miscellaneous [23].
Table No. 5: Goals of impurity investigations
Process-related impurities Degradation–related impurities
Identify significant impurities
Determine origin of impurities and method for elimination or reduction
Establish a control system for impurities involving:
1) Processing/manufacturing conditions 2)Suitable analytical methods/ specifications
Identify potential degradation product through stress testing and actual degradation products through stability studies.
Understand degradation pathway and methods to minimize degradation.
Establish a control system for impurities involving: 1) Processing/manufacturing conditions 2) Suitable analytical methods/ specifications 3) Long term storage conditions including packaging 4) Formulation.
CONCLUSION
T
his review provides a perspective on impurities in drug substance and drug product. This article provides the valuable information about the impurities source, types and its classification, techniques of isolation and characterization, analytical techniques for the determination, qualification of impurities and critical factors to be considered while preparation of the bulk drugs. Nowadays, it is mandatory requirements in various pharmacopoeias to know the impurities present in API’s. Isolation and characterization of impurities is required for acquiring and evaluating data that establishes biological safety which reveals the need and scope of impurity profiling of drugs in pharmaceutical research. To isolate and quantify the impurities, various instrumental analytical techniques are routinely been used. Isolation and characterization of impurities is mandatory for acquiring and evaluating data that establishes biological safety, which reveals the need and scope for impurity profiling of drugs in pharmaceutical research. To isolate and quantify the impurities, various instrumental analytical techniques have been used. Qualification of the impurities is the process of acquiring and evaluating data that establishes biological safety of an individual impurity; thus, revealing the need and scope of impurityprofiling of drugs in pharmaceutical research. This project is an attempt to understand the concept of impurity profile and various aspects and techniques related to it.
REFERENCES:
1. S. Ahuja, K. Alsante (Eds.). Handbook of Isolation and Characterization of Impurities in Pharmaceuticals, Academic Press, Amsterdam, vol. 5, 2003.
2. S. Gorog (Ed.). Identification and Determination of Impurities in Drugs, Elsevier, Amsterdam, vol. 4, 2000.
3. Ahuja S. Impurities Evaluation of Pharmaceuticals, Marcel Dekker, New York: 1998;1.
4. European Pharmacopoeia, 4th Edition, Council of Europe, Strasbourg, France, 2004.
5. United States Pharmacopeia 29, USP Convention Inc., Rockville, MD, USA, 2006.
6. The Japanese Pharmacopoeia, 14th Edition, Society of Japanese Pharmacopoeia, Tokyo, Japan, 2001.
8. Chatwal GR. Pharmaceutical Inorganic chemistry, Himalaya, New Delhi, Vol.1, 1991.
9. Qiu F, Norwood DL. Identification of Pharmaceutical Impurities. J liq Chromatogr & Rel Tech 2007;30:877-935.
10. Shah SR, Patel MA, Naik MV, Pradhan PK, Upadhyay UM. Recent Approaches of Impurity Profiling in Pharmaceutical Analysis: A Review. Int J Pharma Sci and Res 2012;3(10):3603-3617. 11. Roy J. Pharmaceutical Impurities- A Mini review. AAPS Pharm
Sci Tech 2002;3(2):1-8.
12. Patil P, Dr. Vaidya I. Overview on Impurity Profiling. Int J for Pharma Res Schol 2013;2(2):54-65.
13. Solanki R. Impurity profiling of Active Pharmaceutical Ingredients and Finished drug products. Int J Res and Tech 2012;2(3):231-238.
14. Lakshmana Prabu S, Suriyaprakash TNK. Impurities and Its Importance in Pharmacy. Int J Pharma Sci Rev and Res 2010;3(2):66-71.
15. Vijaylakshmi R, Kumaravel S, Anbazhagan S. Scientific Approaches for Impurity profiling in New Pharmaceutical Substances and its products- An Overview. Int J Pharma and Chem Sci 2012.
16. Sapra A, Kakkar S, Narasimhan B. Sources of impurities: A Review. Int Res J Pharma 2012;3(1):57-59.
17. Federal Register, International Conferences on Harmonization. Guidance for Industry: Impurities Residual Solvents, US Department of Health and Human Services Food and Drug Administration, (CDER), Q3C, 1997;1-13.
18. International Conference on Harmonisation (ICH) Guidelines, Q3C and Q3C(M): Impurities: Guideline for Residual Solvents, July 1997 and September 2002, respectively
19. Riley TN. Steric aspects of drug action. Pharmacist. 1998;23:40– 51.
20. Hatakka H, Alatalo H, Palosaari S. Effect of Impurities and additives on crystal Growth. [Last accessed on 2010]. Available from: http://www2.lut.fi/~hhatakka/docit/impure.html 21. Basak AK, Raw AS, Al Hakim AH, Furness S, Samaan NI, Gill DS et
al. Pharmaceutical impurities: Regulatory perspective for Abbreviated New Drug Applications. Adv Drug Deli Reviews, 2007;59:64-72.
22. U.S. Food and Drug Administration. Guidance for Industry, Q3A Impurities in New Drug Substances. February 2003.
23. U.S. Food and Drug Administration. Guidance for Industry, Q3B Impurities in New Drug Products. July 2006.
How to cite this article:
Divya Yada et al. IMPURITY PROFILING: AN EMERGING TASK TO PHARMACEUTICAL INDUSTRIES NOW A DAYS. J Sci Res Pharm 2017;6(10):117-124.
Conflict of interest: The authors have declared that no conflict of interest exists.