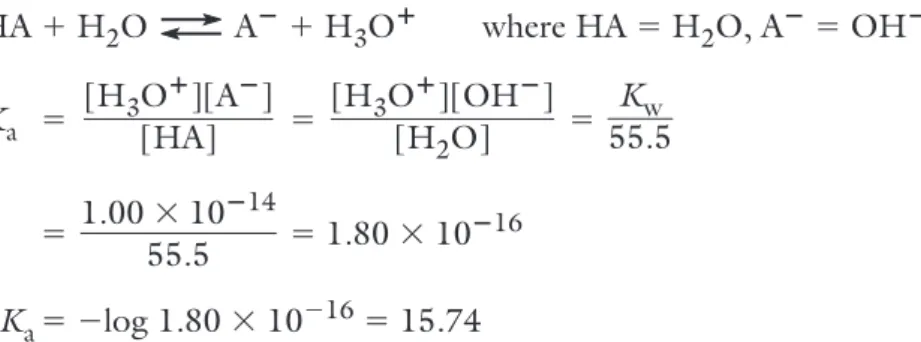1.1 Functional Groups in Biological Chemistry 1.2 Acids and Bases; Electrophiles and Nucleophiles
Brønsted–Lowry Acids and Bases Lewis Acids and Bases
Electrophiles and Nucleophiles
1
1
Common Mechanisms in
Biological Chemistry
This scheme shows the active site of the enzyme that catalyzes the transamination reaction in the biosynthesis of phenylalanine from chorismate. A mechanistic understanding of biosynthetic path-ways is a powerful tool for elucidating the chemical logic of living systems.
1.3 Mechanisms: Electrophilic Addition Reactions 1.4 Mechanisms: Nucleophilic Substitution Reactions 1.5 Mechanisms: Nucleophilic Carbonyl Addition Reactions
Nucleophilic Addition Reactions Alcohol Formation
Imine (Schiff Base) Formation Acetal Formation
Conjugate (1,4) Nucleophilic Additions
1.6 Mechanisms: Nucleophilic Acyl Substitution Reactions 1.7 Mechanisms: Carbonyl Condensation Reactions 1.8 Mechanisms: Elimination Reactions
1.9 Oxidations and Reductions Problems
The final decades of the 20th centurysaw the beginning of a scientific revolution. Based on our newly acquired ability to manipulate, sequence, and synthesize deoxyribonucleic acid (DNA), the way is now open to isolate, study, and eventually modify each of the approximately 30,000 genes in our bodies. Medicines will become safer, more effective, and more specific; terrible genetic diseases such as sickle-cell anemia and cystic fibrosis will be cured; life spans will increase and the quality of life will improve as heart disease and cancer are brought under control.
None of these changes could occur without a detailed knowledge of chemistry, for it is our understanding of life processes at the molecular level that has made this revolution possible and that will sustain it. Biochemical processes are not mysteri-ous. It’s true that the many proteins, enzymes, nucleic acids, polysaccharides, and other substances in living organisms are enormously complex, but despite their
complexity, they are still molecules. They are subject to the same chemical laws as all other molecules, and their reactions follow the same rules of reactivity and take place by the same mechanisms as those of far simpler molecules.
The focus of this book is on examining biochemical processes from a chemical perspective. We’ll begin with a brief review of organic chemistry, looking first at the common functional groups found in biological molecules and then at some fundamental mechanisms by which organic molecules react. Following this gen-eral review of organic reactivity, we’ll look at the structures and chemical charac-teristics of the main classes of biomolecules: carbohydrates, lipids, proteins, enzymes, and nucleic acids. Finally, we’ll come to the heart of the matter: the organic chemistry of biological transformations. We’ll dissect the details of important biochemical pathways to see both howand whythese pathways occur. The result will be both a deeper understanding of biochemistry and a deeper appreciation for the remarkable subtleties by which living organisms function.
1.1 Functional Groups in Biological Chemistry
Chemists have learned through experience that organic compounds can be classi-fied into families according to their structural features and that members of a given family have similar chemical reactivity. The structural features that make such classifications possible are called functional groups. Afunctional groupis a group of atoms within a molecule that has a characteristic chemical behavior. Chemically, a functional group behaves in pretty much the same way in every molecule where it occurs. An ester (RCO2R), for instance, usually undergoes a hydrolysis reaction with water to yield a carboxylic acid (RCO
2H) and an alcohol
(ROH); a thiol (RSH) usually undergoes an oxidation reaction to yield a disul-fide (RSSR); and so on. Table 1.1 lists some common functional groups found in biological molecules.
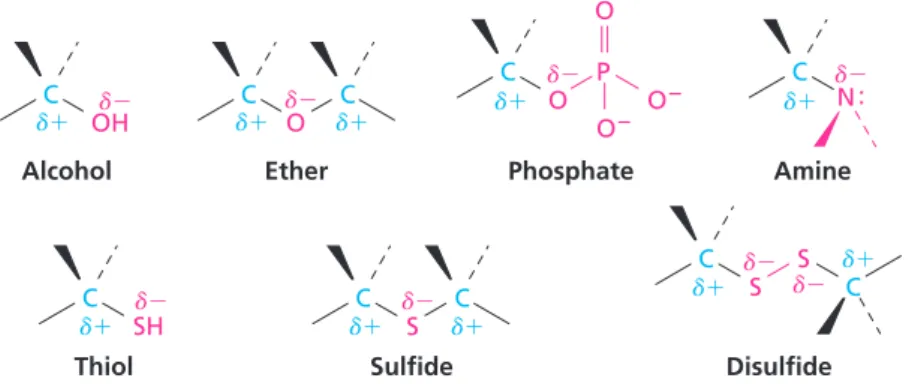
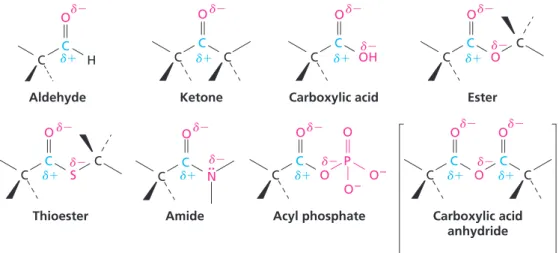
: Carbonyl group C O : : .. C C O N C H C C C C C OH C C O C C C S C C C O Aldehyde Ketone Carboxylic acid Ester Thioester Amide Acyl phosphate C C C N Imine (Schiff base) : C N Diphosphate C O P O O O2 P O O2 O2 O P O O2 O2 Structure* Name Structure* Name Alkene (double bond) Arene (aromatic ring) Alcohol Ether Amine Thiol Sulfide Disulfide Monophosphate
* The bonds whose connections aren’t specified are assumed to be attached to carbon or hydrogen atoms in the rest of the molecule.
C OH C O C C O P O2 O O2 C SH C S C C S C S C C O O O O O
Alcohols, ethers, amines, thiols, sulfides, disulfides, and phosphates all have a carbon atom singly bonded to an electronegative atom. Alcohols, ethers, and phosphates have a carbon atom bonded to oxygen; amines have a carbon atom bonded to nitrogen; and thiols, sulfides, and disulfides have a carbon atom bonded to sulfur. In all cases, the bonds are polar, with the carbon atom being electron-poor and thus bearing a partial positive charge (d1), while the electro-negative atom is electron-rich and thus bears a partial electro-negative charge (d2). These polarity patterns are shown in Figure 1.1.
Note particularly in Table 1.1 the different families of compounds that con-tain the carbonyl group, C O. Carbonyl groups are present in the vast major-ity of biological molecules. These compounds behave similarly in many respects but differ depending on the identity of the atoms bonded to the carbonyl-group carbon. Aldehydes have at least one hydrogen bonded to the CAO; ketones have two carbons bonded to the CAO; carboxylic acids have an OOH group bonded to the CAO; esters have an ether-like oxygen (2OR) bonded to the CAO; thio-esters have a sulfide-like sulfur (2SR) bonded to the CAO; amides have an amine-like nitrogen (2NH2,2NHR, or 2NR2) bonded to the CAO; and acyl phosphates have a phosphate group (2OPO
3
22) bonded to the CAO. You might note that an acyl phosphate is structurally (and chemically) similar to a car-boxylic acid anhydride.
As shown in Figure 1.2, carbonyl compounds are polar, with the electron-poor CAO carbon atom bearing a partial positive charge and the electron-rich oxygen atom bearing a partial negative charge.
1.1 Functional Groups in Biological Chemistry 5
Phosphate Amine Ether
Alcohol
Thiol Sulfide Disulfide
C OH C SH C S C C S C S C O P O2 O O2 : C N C O C d2 d2 d2 d2 d2 d2 d2 d2 d1 d1 d1 d1 d1 d1 d1 d1 d1 d1
FIGURE 1.1 Polarity patterns of some common functional groups. The electronegative atom bears a partial negative charge (d2), and the carbon atom bears a partial positive charge (d1).
1.2 Acids and Bases; Electrophiles and Nucleophiles
Brønsted–Lowry Acids and Bases
Acids and bases are enormously important in biochemistry. The vast majority of biological transformations are catalyzed by acids or bases, and a thorough knowl-edge of acid–base chemistry is crucial for understanding how reactions occur.
According to the Brønsted–Lowry definition, an acidis a substance that donates a proton (hydrogen ion, H1), and a baseis a substance that accepts a proton. A car-boxylic acid such as acetic acid, for example, can donate its OOH proton to a base such as methylamine in a reversible, proton-transfer reaction. The product that results by loss of H1from an acid is the conjugate baseof the acid, and the product that results from addition of H1to a base is the conjugate acidof the base.
.. 1 N C H H H H H H :N C H H H H H C O H H H
(Conjugate base) (Conjugate acid) Methylamine (Base) Acetic acid (Acid) C C O O H H H H + + : .. .. .. : C O : : :2 Acyl phosphate
Thioester Carboxylic acid
anhydride C C S O C Aldehyde C C O H Ketone C C C O Carboxylic acid C C O OH Ester C C O O C Amide .. C C O N C C O O2 O 2 O P O C C O O C C O d2 d2 d2 d2 d2 d2 d2 d2 d2 d2 d2 d2 d2 d2 d2 d1 d1 d1 d1 d1 d1 d1 d1 d1
FIGURE 1.2Polarity patterns in some carbonyl-containing functional groups. The carbonyl carbon atom is electron-poor (d1) and the oxygen atom is electron-rich (d2).
1.2 Acids and Bases; Electrophiles and Nucleophiles 7
Note the standard convention used to show how this proton-transfer reaction occurs: A curved arrow (red) indicates that a pair of electrons moves fromthe atom at the tail of the arrow (the nitrogen in methylamine) tothe atom at the head of the arrow (the acidic hydrogen in acetic acid). That is, the electrons used to form the new NOH bond flow from the base to the acid. As the NOH bond forms, the OOH bond breaks and its electrons remain with oxygen, as shown by a second curved arrow. A curved arrow always represents the movement of electrons, not atoms. Acids differ in their ability to donate protons. Recall from general chemistry that the strength of an acid HA in water solution is expressed by its pK
a, the negative common logarithm of its acidity constant, Ka. A stronger acid has a smaller pK
a(or larger Ka); a weaker acid has a larger pKa(or smaller Ka).
For the reaction: HA 1H2O A21H3O1
K
a5 and pKa5 2logKa Stronger acid—smaller pKa
Weaker acid—larger pKa
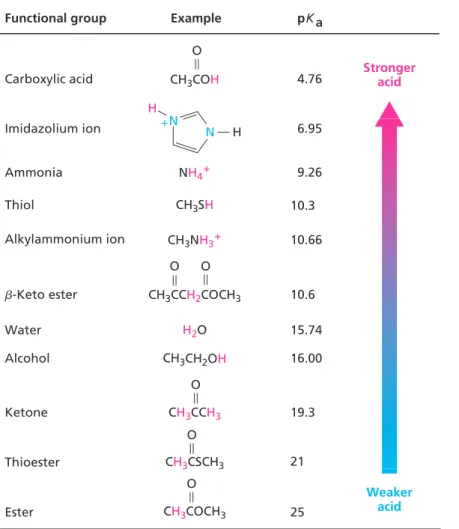
Table 1.2 lists the pK
a’s of some typical acids encountered in biochemistry.
Note that the pKa of water is 15.74, the value that results when Kw, the ion-product constant for water, is divided by the molar concentration of pure water, 55.5 M: HA 1H 2O A21H3O1 where HA 5H2O, A25OH2 Ka 5 5 5 5 51.80 310216 pK a5 2log 1.80 3102 16 515.74
Note also in Table 1.2 that carbonyl compounds are weakly acidic, a point we’ll discuss in more detail in Section 1.7.
1.00 310214 55.5 Kw 55.5 [H3O1][OH2] [H2O] [H3O1][A2] [HA] [H 3O1][A2] [HA]
Table 1.2 Relative Strengths of Some Acids
Just as acids differ in their ability to donate a proton, bases differ in their abil-ity to accept a proton. The strength of a base B in water solution is normally expressed using the acidityof its conjugate acid, BH1.
For the reaction: BH11H2O B1H3O1
Ka5 so Ka3Kb 5q r q r 5[H3O1] [OH2]5Kw51.00 310214 [BH1][OH2] [B] [B][H3O1] [BH1] [B][H3O1] [BH1] 10.3 pKa Example Functional group Stronger acid Weaker acid Carboxylic acid 4.76 Alcohol 16.00 Water 15.74 Ketone 19.3 Imidazolium ion 6.95 Alkylammonium ion 10.66 Ester 25 Thioester 21 b-Keto ester 10.6 Thiol Ammonia 9.26 F O CH3COH N N H H 1 NH41 CH3SH CH3NH31 O O CH3CCH2COCH3 H2O CH3CH2OH O CH3CCH3 O CH3CSCH3 O CH3COCH3
1.2 Acids and Bases; Electrophiles and Nucleophiles 9
Thus Ka5 and Kb5
so pK
a1pKb514 and pKb514 2pKa Stronger base—larger pKafor BH1
Weaker base—smaller pKafor BH1
These equations say that we can determine the basicity of a base B by knowing the K
aof its conjugate acid BH1. A stronger base holds H1more tightly, so it
has a weaker conjugate acid (larger pKa); a weaker base holds H1less tightly, so it has a stronger conjugate acid (smaller pK
a). Table 1.3 lists some typical bases
found in biochemistry. Note that water can act as either a weak acid or a weak base, depending on whether it donates a proton to give OH2or accepts a proton to give H3O1. Similarly with imidazole, alcohols, and carbonyl compounds, which can either donate or accept protons depending on the circumstances.
Table 1.3 Relative Strengths of Some Bases
Kw Ka Kw Kb Alcohol 12.5 Guanidino 6.95 21.74 10.66 9.26 Imidazole Amine Ammonia Water pKa of BH1 Exampl e Functional group Hydroxide ion 15.74 Ketone 27.5 22.05 :N N H :NH3 1NH4 .. CH3NH2 ::NH H2NCNHCH2CH3 1NH 2 H2NCNHCH2CH3 .. : H2O H3O1 .. .. CH3OH :: ::O CH3CCH3 1OH CH3CCH3 Stronger base Weaker base :: .. .. OH2 H2O CH3NH3 1 N N H H 1 CH3OH2 1
Lewis Acids and Bases
The Brønsted–Lowry definition of acids and bases covers only compounds that donate or accept H1. Of more general use, however, is the Lewis definition. A Lewis acidis a substance that accepts an electron pair from a base, and a Lewis baseis a substance that donates an electron pair to an acid. For all practical pur-poses, Lewis and Brønsted–Lowry bases are the same: Both have lone pairs of electrons that they donate to acids. Lewis and Brønsted–Lowry acids, however, arenotnecessarily the same.
The fact that a Lewis acid must be able to accept an electron pair means that it must have a vacant, low-energy orbital. Thus, the Lewis definition of acidity is much broader than the Brønsted–Lowry definition and includes many species in addition to H1. For example, various metal cations and transition-metal com-pounds, such as Mg21, Zn21, and Fe31are Lewis acids.
Lewis acids are involved in a great many biological reactions, often as cofac-tors in enzyme-catalyzed processes. Metal cations such as Mg21 and Zn21are particularly common, but complex compounds such as iron–sulfur clusters are also found. We’ll see an example in Section 4.4 where citrate undergoes acid-catalyzed dehydration to yield cis-aconitate, a reaction in the citric acid cycle.
+ B :: AA Lewis base Filled orbital B : Lewis acid A A Vacant orbital Vacant orbital .. .. : : Fe Fe S FeFe S S FeFe Fe Fe S S Cys S Cys Cys S An iron–sulfur cluster (Cys = cysteine) Fe Fe S FeFe S S FeFe Fe Fe S S H H H O O OH2 B B:: H Fe Fe S FeFe S S FeFe Fe Fe S OH OH
Citrate cis-Aconitate
+ + Cys S Cys S Cys S
Cys Cys S Cys S
OH2 CO22 CO22 CO22 CO22 CO22 CO22 CO22 CO22 CO22 OH Cys SH 1
Electrophiles and Nucleophiles
Closely related to acids and bases are electrophilesand nucleophiles. An electro-phileis a substance that is “electron-loving.” It has a positively polarized, elec-tron-poor atom and can form a bond by accepting a pair of electrons from an electron-rich atom. A nucleophile, by contrast, is “nucleus-loving.” It has a negatively polarized, electron-rich atom and can form a bond by donating a pair of electrons to an electron-poor atom. Thus, electrophiles are essentially the same as Lewis acids and nucleophiles are the same as Lewis bases. In practice, however, the words “acid” and “base” are generally used when electrons are donated to H1 or a metal ion, and the words “electrophile” and “nucleophile” are used when electrons are donated to a carbon atom.
Electrophiles are either positively charged or neutral and have a positively polarized, electron-poor atom that can accept an electron pair from a nucleo-phile/base. Acids (H1donors), trialkylsulfonium compounds (R3S1), and car-bonyl compounds are examples (Figure 1.3).
Nucleophiles are either negatively charged or neutral and have a lone pair of electrons they can donate to an electrophile/acid. Amines, water, hydroxide ion, alkoxide ions (RO2), and thiolate ions (RS2) are examples.
1.2 Acids and Bases; Electrophiles and Nucleophiles 11
O2 C Nu 1 1 1 1 + Nu —HH S H C C C O O2 H H :Nu :Nu :Nu C C C O C H H H H H H H C C C O O H H H H
(a) Acetic acid (b) Trimethylsulfonium ion (c) Acetone
S S Nu — CH3 CH3 CH3 CH3 H3C H3C H3C H3C + d1 d1 d1
1.3 Mechanisms: Electrophilic Addition Reactions
Chemical reactions carried out in living organisms follow the same rules of reac-tivity as reactions carried out in the laboratory. The “solvent” is often different, the temperature is often different, and the catalyst is certainly different, but the reac-tions occur by the same fundamental mechanisms. That’s not to say that all bioor-ganic reactions have obvious laboratory counterparts—some of the most chemi-cally interesting biotransformations cannot be duplicated in the laboratory without an enzyme because too many side reactions would occur. Nevertheless, the chemical mechanisms of biotransformations can be understood and accounted for by organic chemistry. In this and the remaining sections of Chapter 1, we’ll look at some fundamental organic reaction mechanisms, beginning with the elec-trophilic addition reactions of CAC bonds.
An electrophilic addition reactionis initiated by addition of an electrophile to an unsaturated (electron-rich) partner, usually an alkene, and leads to forma-tion of a saturated product. In the laboratory, for example, water undergoes an acid-catalyzed electrophilic addition to 2-methylpropene to yield 2-methyl-2-propanol. The reaction takes place in three steps and proceeds through a posi-tively charged, carbocation intermediate (Figure 1.4). In the first step, electrons from the nucleophilic CAC bond attack an electrophilic hydrogen atom of H
3O1, forming a COH bond. The intermediate carbocation then reacts with
water as nucleophile, giving first a protonated alcohol and then the neutral alco-hol after a proton-transfer step that regenerates H
3O1. Note that the initial
pro-tonation takes place on the less highly substituted carbon of the double bond, leading to the more highly substituted, more stable, carbocation.
Biological examples of electrophilic addition reactions occur frequently in the biosynthetic routes leading to steroids and other terpenoids, although they are less common elsewhere. The electrophile in such reactions is a positively charged or positively polarized carbon atom, which often adds to a CAC bond within the same molecule. As an example, a-terpineol, a substance found in pine oil and used in perfumery, is derived biosynthetically from linalyl diphos-phate by an internal electrophilic addition reaction. Following formation of an allylic carbocation by dissociation of the diphosphate (here abbreviated PPO),
electrophilic addition to the nucleophilic CAC bond at the other end of the molecule occurs, giving a second carbocation that then reacts with nucleophilic water. A proton transfer from the protonated alcohol to water yields a-terpineol (Figure 1.5). We’ll see more such examples when we look at steroid biosynthe-sis in Section 3.5.
1.3 Mechanisms: Electrophilic Addition Reactions 13
:.. C C H H H O H H C C H H H : : H O H Carbocation C C H H H HO OH2 C C H H + H O H H
The electrophile H3O1is attacked by the nucleophilic double bond, forming a C—H bond and giving a carbocation intermediate. H3C H3C H3C H3C H3C H3C H3C H3C 1 1 1
The nucleophile H2O attacks the electrophilic carbocation, forming a C—O bond.
A proton-transfer reaction with water regenerates H3O1and yields the alcohol product.
H3O1
FIGURE 1.4 The mechanism of the acid-catalyzed electrophilic addition of water to 2-methyl-propene. The reaction involves a carbocation intermediate.
1.4 Mechanisms: Nucleophilic Substitution Reactions
Anucleophilic substitution reactionis the substitution of one nucleophile (the
leaving group) by another on a saturated, sp3-hybridized carbon atom: Br2 by OH2, for example. Nucleophilic substitution reactions in the laboratory gener-ally proceed by either an SN1 mechanismor an SN2 mechanismdepending on the reactants, the solvent, the pH, and other variables. S
N1 reactions usually take
place with tertiary or allylic substrates and occur in two steps through a carboca-tion intermediate. S
N2 reactions usually take place with primary substrates and
take place in a single step without an intermediate. The mechanism of a typical S
N1 reaction is shown in Figure 1.6. As indicated,
the substrate undergoes a spontaneous dissociation to generate a carbocation intermediate, which reacts with the substituting nucleophile to give product.
The mechanism of a typical SN2 process is shown in Figure 1.7 for the reac-tion of hydroxide ion with (S)-2-bromobutane. The reaction takes place in a sin-gle step when the incoming nucleophile uses a lone pair of electrons to attack the
Linalyl diphosphate a-Terpineol PPO OH + PPO = 2O P O O P O O2 O2 O H H O .. : CH3 CH3 CH3 CH3 OH2 .. :OH2 CH2 CH3 H3C H3C H3C H3C H3C H3C H3C CH3 CH3 CH3 1 1 1 H3O1
FIGURE 1.5 The biosynthesis of a-terpineol from linalyl diphosphate occurs by an electrophilic addi-tion reacaddi-tion.
electrophilic carbon atom of the alkyl halide from a direction 180° opposite the COBr bond. As the OH2comes in and a new OOC bond begins to form, the old COBr bond begins to break and the Br2leaves. Because the incoming and outgoing nucleophiles are on opposite sides of the molecule, the stereochemistry at the reacting center inverts during an S
N2 reaction. (S)-2-Bromobutane yields
(R)-2-butanol, for example. (There is no guarantee that inversion will change the assignment of a stereocenter from Rto Sor vice versa, because the relative pri-orities of the four groups attached to the stereocenter may also change.)
1.4 Mechanisms: Nucleophilic Substitution Reactions 15
Carbocation
C O
H
H
Spontaneous dissociation of the substrate occurs to generate a carbocation intermediate plus bromide ion. C OH C Br C + + Br2 H3C H3C H3C H3C H3C H3C H3C H3C H3C CH3 CH3 CH3 .. :OH2 .. :OH2 1 1
The carbocation reacts with water nucleophile to yield a protonated alcohol intermediate...
...which undergoes a proton transfer to water to give the final substitution product.
H3O1
FIGURE 1.6 Mechanism of the SN1 reaction of 2-bromo-2-methylpropane with water to yield 2-methyl-2-propanol. The reaction occurs by a spontaneous dissociation to give a carbocation intermediate, which reacts with water.
Biochemical examples of both S
N1 and SN2 reactions occur in numerous
path-ways. An SN1 reaction, for instance, takes place during the biological conversion of geranyl diphosphate to geraniol, a fragrant alcohol found in roses and used in per-fumery. Initial dissociation of the diphosphate gives a stable allylic carbocation, which reacts with water nucleophile and transfers a proton to yield geraniol.
An S
N2 reaction is involved in biological methylation reactions whereby a
OCH3group is transferred from S-adenosylmethionine to various nucleophiles.
An SN1 reaction Geranyl diphosphate Geraniol OPP + + CH2 CH2OH2 CH2OH CH2 .. :OH2 1 H3O1 H2O C C + HO HO HO HO Br Br Br2 Br2 C C H H .. .. .. ..:: CC Br Br H d2 d2 dd22 CH3CH2 CH3CH2 CH2CH3 HO2 H3C CH3 CH3
OH2nucleophile uses a lone pair of electrons to attack the electrophilic alkyl halide carbon from a direction 1808oppoite the C—Br bond. The transition state has partially formed C—O and C—Br bonds.
The stereochemistry at carbon inverts as the C—H bond forms fully and the Br2ion leaves.
FIGURE 1.7 Mechanism of the SN2 reaction of (S)-2-bromobutane with hydroxide ion to yield (R)-2-butanol. The reaction occurs in a single step with inversion of stereochemistry at the reacting carbon atom.
In the biosynthetic transformation of norepinephrine to epinephrine (adrena-line), for instance, the nucleophilic amine nitrogen atom of norepinephrine attacks the electrophilic methyl carbon atom of S-adenosylmethionine in an S
N2
reaction, displacing S-adenosylhomocysteine as the leaving group (Figure 1.8).
1.5 Mechanisms: Nucleophilic Carbonyl Addition Reactions
Carbonyl groups are present in the vast majority of biological molecules, and car-bonyl reactions are thus encountered in almost all biochemical pathways. In dis-cussing carbonyl-group chemistry, it’s useful to make a distinction between two general classes of compounds. In one class are aldehydes and ketones, which have their carbonyl carbon bonded to atoms (C and H) that can’t stabilize a negative charge and therefore don’t typically act as leaving groups in substitution reac-tions. In the second class are carboxylic acids and their derivatives, which have
1.5 Mechanisms: Nucleophilic Carbonyl Addition Reactions 17
Norepinephrine Epinephrine (Adrenaline) S-Adenosylmethionine S-Adenosylhomocysteine + + :: HO HO HO H O S N OH OH N N N HO HO HO H O S N OH OH N N N An SN2 reaction NHCH3 CH3 NH3 NH2 NH2 H2N 2O 2C 2O 2C H 1 NH3 H 1 1
FIGURE 1.8 The biosynthesis of epinephrine from norepinephrine occurs by an SN2 reaction with
their carbonyl carbon bonded to an electronegative atom (O, N, or S) that can
stabilize a negative charge and thus canact as a leaving group in a substitution reaction.
The CAO bond in every carbonyl compound, regardless of structure, is polarized with the oxygen negative and the carbon positive. As a result, the car-bonyl oxygen is nucleophilicand reacts with acids/electrophiles, while the carbon is electrophilic and reacts with bases/nucleophiles. These simple reactivity pat-terns show up in almost all biological pathways.
Nucleophilic Addition Reactions
Anucleophilic addition reactionis the addition of a nucleophile (:Nu) to the elec-trophilic carbon of an aldehyde or ketone. As an electron pair from the nucleophile forms a bond to the carbon, an electron pair from the CAO bond moves toward
Electrophilic carbon reacts with bases and nucleophiles.
Nucleophilic oxygen reacts with acids and electrophiles.
C O :: :: F d2 d1 .. C C O N Ketone Aldehyde C C C O C O H Acyl phosphate Thioester C C O O O2 O2 O P C C O S C Amide Ester Carboxylic acid C C O O C C C O OH
No leaving group is attached to the carbonyl carbon, so a substitution reaction is difficult.
A potential leaving group
is attached to the carbonyl carbon, so a substitution reaction ispossible.
oxygen, giving an alkoxide ion, RO2. The carbonyl carbon rehybridizes from sp2to
sp3during the process, so the alkoxide product has tetrahedral geometry.
Once formed, the tetrahedral alkoxide ion can do any of several things, as shown in Figure 1.9. When a nucleophile such as hydride ion (H:2) or a carbanion (R
3C:2) adds, the alkoxide ion undergoes protonation to yield a stable alcohol.
A nucleophilic
addition reaction C :Nu
O :: :: d2 d1 C Nu ..2 O :: ::
1.5 Mechanisms: Nucleophilic Carbonyl Addition Reactions 19
OH C NHR Carbinolamine OH C H C NR OR C OR H Alcohol Imine (Schiff base) Acetal OH C OR Hemiacetal ROH 1 1 1 C H C H R O C H H R N + :: Nu = :: H2 (a) :: Nu = RNH2 .. (b) :: Nu = ROH .. .. (c) F :Nu C O :: :: d2 d1 C Nu ..2 O :: :: ..2 O :: :: :: ::O..2 :: ::O..2 H2O + H2O
FIGURE 1.9 Some typical nucleophilic addition reactions of aldehydes and ketones. (a) With a hydride ion as nucleophile, protonation of the alkoxide intermediate leads to an alcohol. (b) With an amine as nucleophile, proton transfer and loss of water leads to an imine. (c) With an alcohol as nucleophile, proton transfer leads to a hemiacetal, and further reaction with a second equivalent of alcohol leads to an acetal.
When a primary amine nucleophile (RNH
2) adds, the alkoxide ion undergoes a
pro-ton transfer to yield a carbinolamine, which loses water to form an imine (R
2CANR9), often called a Schiff basein biochemistry. When an alcohol
nucleo-phile (ROH) adds, the alkoxide undergoes proton transfer to yield a hemiacetal, which can react with a second equivalent of alcohol and lose water to give an acetal [R2C(OR9)2]. In all the reactions that follow, note the role of acid and base catalysts.
Alcohol Formation
In the laboratory, the conversion of an aldehyde or ketone to an alcohol is gener-ally carried out using NaBH4as the nucleophilic hydride-ion donor. In biological pathways, however, NADH (reduced nicotinamide adenine dinucleotide) or the closely related NADPH (reduced nicotinamide adenine dinucleotide phosphate) is the most frequently used hydride-ion donor. An example that occurs in the pathway by which organisms synthesize fatty acids is the conversion of aceto-acetyl ACP (acyl carrier protein) to 3-hydroxybutyryl ACP. We’ll look at the details of the process in Section 3.4.
Imine (Schiff Base) Formation
Imines are formed in a reversible, acid-catalyzed process that begins with nucleo-philic addition of a primary amine to the carbonyl group of an aldehyde or ketone. The initial dipolar addition product undergoes a rapid proton transfer that removes an H1 from N and places another H1 on O to give a carbinolamine, which is
C C C O SACP H H H O .. H H C O N H— A
Acetoacetyl ACP NADPH
3-Hydroxybutyryl ACP + + C C C O SACP H H H OH C O C C O SACP H H NH2 H3C H3C H3C 2 NADP1 C H N O NH2 1
protonated on the oxygen atom by an acid catalyst. The effect of this protonation is to convert OOH into a much better leaving group (OOH21) so that it can be expelled by the electrons on nitrogen. Deprotonation of the resultant iminium ion then gives the imine product. The mechanism is shown in Figure 1.10.
1.5 Mechanisms: Nucleophilic Carbonyl Addition Reactions 21
+ H2O H—A + H—A A C N—R C NHR C C NHR C O : : : : ::
Nucleophilic attack on the carbonyl group by the amine leads to a dipolar intermediate.
Proton transfer occurs by removing a proton from N and adding a proton to O, giving a neutral carbinolamine.
An acid catalyst H—A protonates the oxygen to make the —OH a better leaving group.
Lone-pair electrons from nitrogen expel water, giving an iminium ion.
Deprotonation of the iminium ion regenerates the acid catalyst and yields the imine product.
Carbinolamine Imine Iminium ion O : : : N:HR OH : OH2 C NH2R NH2R 2 1 1 1 2
FIGURE 1.10 Mechanism of acid-catalyzed imine (Schiff base) formation by reaction of an aldehyde or ketone with a primary amine, RNH2.
The conversion of a ketone to an imine is a step in numerous biological path-ways, including the route by which many amino acids are synthesized in the body. For instance, the ketone pyruvate and the amine pyridoxamine phosphate, a de-rivative of vitamin B6, form an imine that is converted to the amino acid alanine. We’ll look at the details in Section 5.1.
Acetal Formation
Acetals, like imines, are formed in a reversible, acid-catalyzed process. The reac-tion begins with protonareac-tion of the carbonyl oxygen to increase its reactivity, fol-lowed by nucleophilic addition of an alcohol. Deprotonation then gives a neutral hemiacetal, and reprotonation on the hydroxyl oxygen converts the OOH into a better leaving group so that it can be expelled by electrons on the neighboring
OOR to produce an oxonium ion. This oxonium ion behaves much like the pro-tonated carbonyl group in the first step, undergoing a second nucleophilic addi-tion with alcohol. A final deprotonaaddi-tion then gives the acetal product. The mechanism is shown in Figure 1.11.
+ + An imine C O Pyruvate Pyridoxamine phosphate 1 1 N OH H C H H N OH H C N C H H 22O 3POCH2 22O3POCH2 CO22 CO22 H3C CH3 CH3 CH3 NH2 H2O
1.5 Mechanisms: Nucleophilic Carbonyl Addition Reactions 23
H—A
Nucleophilic addition of alcohol occurs, and deprotonation gives a hemiacetal.
A2 + HA
...addition of a second alcohol molecule. C Hemiacetal C 1 1 C O H R C + C H R
Protonation of the oxygen polarizes the carbonyl group and makes it a better acceptor.
Protonation of the hemiacetal hydroxyl activates the —OH for expulsion.
Deprotonation then regenerates the acid catalyst and gives the acetal. Loss of water yields an oxonium ion, which is activated for...
C R— —H C + H—A C Acetal F H2O A :OH 1 :OH2 1 :OR :OR :O:H :O:H : 1 O: : : O R— —H: : : :O:R O : : OR : : OR : : OR : :O 2
FIGURE 1.11 Mechanism of acid-catalyzed hemiacetal and acetal formation by reaction of an alde-hyde or ketone with an alcohol.
The formation of hemiacetals and acetals is a central part of carbohydrate chemistry. Glucose, for instance, is in a readily reversible equilibrium between open (aldehyde 1alcohol) and closed (cyclic hemiacetal) forms. Many glucose molecules can then join together by acetal links to form starch and cellulose (Sec-tion 4.1).
Conjugate (1,4) Nucleophilic Additions
Closely related to the direct (1,2) addition of a nucleophile to the CAO bond of an aldehyde or ketone is the conjugate (1,4) addition, or Michael reaction, of a nucleophile to the CAC bond of an a,b-unsaturated aldehyde or ketone (or thioester). The initial product, a resonance-stabilized enolate ion, typically undergoes protonation on the acarbon (the carbon next tothe CAO) to give a saturated aldehyde or ketone (or thioester) product.
Glucose (open) O HO HO OH O HO HO OH C C H H H O C C OH OH OH OH OH C HO H C H b-Glucose (hemiacetal) a-Glucose (hemiacetal) F HOCH2 CH2OH HOCH2 2 1 C C C O H C C C Nu HO C C C O2 Nu Nu2 A direct (1,2) addition 1 :
Note that this conjugate addition reaction gives an overall result similar to that of the electrophilic alkene addition discussed in Section 1.3, but the mechanisms of the two processes are entirely different. Isolated alkenes react with electrophiles
and form carbocation intermediates; a,b-unsaturated carbonyl compounds react with nucleophilesand form enolate-ion intermediates. Conjugate addition occurs because the electronegative oxygen atom of the a,b-unsaturated carbonyl com-pound withdraws electrons from the bcarbon, thereby making it more electron-poor and more electrophilic than a typical alkene CAC bond.
1.5 Mechanisms: Nucleophilic Carbonyl Addition Reactions 25
2 1 4 3 b a C C O C C Nu C C C C C C C C Nu C C O C C Nu H H1 A conjugate (1,4) addition (Michael reaction) Saturated aldehyde/ketone a,b-Unsaturated aldehyde/ketone Enolate ion : :O: : :Nu2 2 2 : :O C C C O 1 1 2 2 C C C O A saturated ketone: An a,b-unsaturated ketone: Electrophilic C C O C C C C O C C
Among the many biological examples of conjugate additions is the conversion of fumarate to malate by reaction with water, a step in the citric acid cycle by which acetate is metabolized to CO
2.
1.6 Mechanisms: Nucleophilic Acyl Substitution Reactions
Carboxylic acids and their derivatives are characterized by the presence of an elec-tronegative atom (O, N, S) bonded to the carbonyl carbon. As a result, these compounds can undergo nucleophilic acyl substitution reactions—the substi-tution of the leaving group bonded to the carbonyl carbon (:Y) by an attacking nucleophile (:Nu2).
As shown in Figure 1.12 for the reaction of OH2 with methyl acetate, a nucleophilic acyl substitution reaction is initiated by addition of the nucleophile to the carbonyl carbon in the usual way. But the tetrahedrally hybridized alkoxide intermediate is not isolated; instead, it reacts further by expelling the leaving group and forming a new carbonyl compound. The overall result of nucleophilic acyl substitution is the replacement of the leaving group by the attacking nucleo-phile, just as occurs in the nucleophilic alkyl substitutions discussed in Section 1.4. Only the mechanismsof the two substitutions are different.
Both the addition step and the elimination step can affect the overall rate of a nucleophilic acyl substitution reaction, but the first step is generally rate-limiting. Thus, the greater the stability of the carbonyl compound, the less reactive it is. We therefore find that, of the carbonyl-containing functional groups commonly
H H OH H H O H H H H—A Malate Fumarate B H H OH : : 2O 2C 2O2C 2O2C CO22 CO22 CO2 2 2 A nucleophilic acyl substitution reaction : : : Nu2 + + Y:2 C Y O : : C Nu O
found in living organisms, amides are the least reactive because of resonance sta-bilization; esters are somewhat more reactive; and thioesters and acyl phosphates are the most reactive toward substitution. (Acyl phosphates are generally further activated for substitution by complexation with a Lewis acidic metal cation such as Mg21.) Ester C C O O C .. C C O N Amide Thioester C C O S C C C O O P O O2 O2 Acyl phosphate F Reactivity
1.6 Mechanisms: Nucleophilic Acyl Substitution Reactions 27
+ +
A subsequent acid–base reaction deprotonates the carboxylic acid.
:OH2
Nucleophilic addition of OH2 to the ester yields a tetrahedral alkoxide-ion intermediate. C O O C OH C O2 C OH
An electron pair from the alkoxide oxygen moves toward carbon, regenerating a CAO bond and expelling CH3O2as leaving group.
.. .. : :O.. F : : O : : O : : CH3 CH3O CH3O2 CH3OH H3C H3C H3C H3C 2
FIGURE 1.12 Mechanism of the nucleophilic acyl substitution reaction of OH2with methyl acetate to give acetate. The reaction occurs by a nucleophilic addition to the carbonyl group, followed by expulsion of the leaving group in a second step.
Nucleophilic acyl substitution reactions occur frequently in biochemistry. For example, the carboxypeptidase-catalyzed hydrolysis of the C-terminal amide bond in proteins is one such process.
1.7 Mechanisms: Carbonyl Condensation Reactions
The third major reaction of carbonyl compounds, carbonyl condensation, occurs when two carbonyl compounds join to give a single product. When an aldehyde is treated with base, for example, two molecules combine to yield a b -hydroxy aldehyde product. Similarly, when an ester is treated with base, two molecules combine to yield a b-keto ester product.
Carbonyl condensation results in bond formation between the carbonyl car-bon of one partner and the acarbon of the other partner. The reactions occur because the ahydrogen of a carbonyl compound is weakly acidic and can there-fore be removed by reaction with a base to yield an enolate ion. Like other anions, enolate ions are nucleophiles.
C O C H O C H O +
Two aldehydes A b-hydroxy aldehyde HO H H O a b C O O O + +
Two esters A b-keto ester
:: Base :: Base a b H3C H3C H3C H3C OCH3 CH3OH OCH3 OCH3 H A C C O2 O N C H R H HO B H O H C C O2 O N C R9 R9 R9 H R H + C O OH C C O N H R H H : : : O :O: :2 O2
The acidity of carbonyl compounds is due to resonance stabilization of the enolate ion, which allows the negative charge to be shared by the acarbon and the electronegative carbonyl oxygen. As shown in Table 1.4, aldehydes and ketones are the most acidic monocarbonyl compounds, with thioesters, esters,
a A carbonyl compound :: Base An enolate ion C C H C C C C : : O :O: :O: : : 2 2
1.7 Mechanisms: Carbonyl Condensation Reactions 29
pKa Example Carbonyl compound Stronger acid Weaker acid Carboxylic acid 4.7 Aldehyde Ketone 17 1,3-Diketone 1,3-Diester 9.0 Ester 25 Thioester 21 b-Keto ester 10.6 12.9 19.3 O CH3COH O O CH3CCH2COCH3 O O CH3OCCH2COCH3 O O CH3CCH2CCH3 O CH3CH O CH3CCH3 O CH3CSCH3 O CH3COCH3 Amide 30 O CH3CN(CH3)2
and amides less so. Most acidic of all, however, are 1,3-dicarbonyl compounds (b-dicarbonyl compounds), which have an aposition flanked by twoadjacent car-bonyl groups.
The condensation of an aldehyde or ketone is called the aldol reactionand takes place by the mechanism shown in Figure 1.13. One molecule reacts with base to give a nucleophilic enolate ion, which then adds to the second molecule in a nucleophilic addition reaction. Protonation of the initially formed alkoxide ion yields the neutral b-hydroxy aldehyde or ketone product. Note that the con-densation is reversible: A b-hydroxy aldehyde or ketone can fragment on treat-ment with base to yield two molecules of aldehyde or ketone.
H C H C O H + C C O H H H H B: : C C O H H H H H C C C C O H H H H H H OH H C C C C O H H H H H H O2
H—A The alkoxide ion is protonated toyield the final b-hydroxy carbonyl product.
...which adds as a nucleophile to the carbonyl group of a second aldehyde molecule, producing an alkoxide ion.
Base deprotonates the aldehyde to give an enolate ion...
Ab-hydroxy carbonyl compound
2
FIGURE 1.13 Mechanism of the aldol reaction, a reversible, base-catalyzed condensation reaction between two molecules of aldehyde or ketone to yield a b-hydroxy carbonyl compound. The key step is nucleophilic addition of an enolate ion to a CAO bond.
1.7 Mechanisms: Carbonyl Condensation Reactions 31
The condensation of an ester is called the Claisen condensation reaction and takes place by the mechanism shown in Figure 1.14. One molecule reacts with base to give a nucleophilic enolate ion, which adds to the second molecule in a nucleophilic acyl substitution reaction. The initially formed alkoxide ion
Ab-keto ester + .. H C H C O + C C O H H H C C O H H H H C C C C O H H H H O H C C C C O H H H H O
...which adds as a nucleophile to the carbonyl group of a second ester molecule, producing an alkoxide ion.
Base deprotonates the ester to give an enolate ion...
The alkoxide ion expels CH3O2 as the leaving group to yield the b-keto ester. H C C C C O H H O H +
Deprotonation of the acidic b-keto ester gives an anion, serving to drive the overall reaction toward completion.
F B: OCH3 OCH3 CH3O CH3OH CH3O2 CH3O2 OCH3 OCH3 OCH3 OCH3 : : : 2 : 2 2
FIGURE 1.14 Mechanism of the Claisen condensation reaction, a reversible, base-catalyzed condensa-tion reaccondensa-tion between two molecules of ester to yield a b-keto ester. The key step is nucleophilic acyl substitution by an enolate ion, with expulsion of an alkoxide leaving group.
expels CH3O2as the leaving group to regenerate a CAO bond and form the
b-keto ester product. As with the aldol reaction, the Claisen condensation is reversible: A b-keto ester can fragment on treatment with base to yield two mole-cules of ester.
Carbonyl condensation reactions are involved in nearly all biochemical path-ways and serve as the primary biological method for forming and breaking car-bon–carbon bonds. For instance, one step in the biosynthesis of glucose from pyruvate is the aldol reaction of glyceraldehyde 3-phosphate with dihydroxyace-tone phosphate. As another example, the biological pathway for terpenoid and steroid biosynthesis begins with a Claisen condensation of the thioester acetyl CoA (coenzyme A) to give acetoacetyl CoA. We’ll look at the details of glucose biosynthesis in Section 4.5 and the details of steroid biosynthesis in Section 3.6.
1.8 Mechanisms: Elimination Reactions
The elimination of HX to yield an alkene appears to be the simple reverse of an electrophilic addition of HX. In fact, though, elimination reactionsare a good deal more complex than additions and can occur by any of several mechanisms. In the laboratory, the three most common processes are the E1, E2, and E1cB reactions, which differ in the timing of COH and COX bond-breaking. In the E1 reaction, the COX bond breaks first to give a carbocation intermediate, which then undergoes base abstraction of H1 to yield the alkene (the exact reverse of the electrophilic addition reaction described in Section 1.3). In the E2
Aldol reaction
Fructose 1,6-bisphosphate
Claisen condensation reaction
Acetyl CoA Acetoacetyl CoA Glyceraldehyde 3-phosphate Dihydroxyacetone phosphate + HSCoA OH OH O + CH3C CH2CSCoA CH3CSCoA O O O O CH3CSCoA + 22O 3POCH2CHCH OH OH 22O 3POCH2CHCH CH2CCH2OPO322 O OH CHCCH2OPO322 O
reaction, base-induced COH bond cleavage is simultaneous with COX bond cleavage, giving the alkene in a single step. In the E1cB reaction (cB for “conju-gate base”), base abstraction of the proton occurs first, giving a carbanion inter-mediate that undergoes loss of X2in a subsequent step to give the alkene.
Examples of all three mechanisms occur in different biological pathways, but the E1cB mechanism is particularly common. The substrate is usually an alcohol (X 5OH) or protonated alcohol (X 5OH
21), and the H atom that is removed
is usually made acidic, particularly in E1cB reactions, by being adjacent to a car-bonyl group. Thus, b-hydroxy carbonyl compounds (aldol reaction products) are frequently converted to a,b-unsaturated carbonyl compounds by elimination
1.8 Mechanisms: Elimination Reactions 33
Carbocation
E1 Reaction: C–X bond breaks first to give a carbocation intermediate, followed by base removal of a proton to yield the alkene.
C C + X2 + B H C C H B: C C H X 1 1
E2 Reaction: C–H and C–X bonds break simultaneously, giving the alkene in a single step without intermediates.
C C H X B: + B—H + X2 C C 1 C C X
E1cB Reaction: C–H bond breaks first, giving a carbanion intermediate that loses X2to form the alkene.
Carbanion F C C H X B: + B—H + X2 C C 1 2:
reactions. An example is the dehydration of a b-hydroxy thioester to the corre-sponding unsaturated thioester, a reaction that occurs in fatty-acid biosynthesis (Section 3.4). The base in this reaction is a histidine residue in the enzyme, and the elimination is assisted by complexation of the OOH group to the protonated histidine as a Lewis acid. Note that the reaction occurs with synstereochemistry, meaning that the OH and OOH groups in this example are eliminated from the same side of the molecule.
1.9 Oxidations and Reductions
Oxidation–reduction, or redox, chemistry is a large and complicated, but extremely important, topic. Rather than attempt a complete catalog of the subject at this point, however, let’s focus on the mechanisms of two of the more commonly occurring biological redox processes: the oxidation of an alcohol and the reduction of a carbonyl compound. We’ll look at the mechanisms of other kinds of biological oxidations in later chapters as the need arises when we discuss specific pathways.
In the laboratory, alcohol oxidations generally occur through a mechanism that involves attachment to oxygen of a leaving group X, usually a metal in a high oxidation state such as Cr(VI) or Mn(VII). An E2-like elimination reaction then forms the CAO bond and expels the metal in a lower oxidation state. Note that the COH hydrogen is removed by base as H1during the elimination step.
Although a similar mechanism does occasionally occur in biological pathways, many biological oxidations of an alcohol occur by a reversible hydride-transfer
C H O H C O C H O X B: C H C C O SR H C C C O SR H H HO H C C C O2 SR H H HO N N His H H N N His H : Unsaturated thioester b -Hydroxy-thioester F H2O H3C H3C H3C 1
mechanism involving one of the coenzymes NAD1(oxidized nicotinamide ade-nine dinucleotide) or NADP1 (oxidized nicotinamide adenine dinucleotide phosphate). As shown in Figure 1.15, the reaction occurs in a single step without intermediates when a base B: abstracts the acidic OOH proton, the electrons from the OOH bond move to form a CAO bond, and the hydrogen attached to carbon is transferred to NAD1. Note that the COH hydrogen is transferred as H2, in contrast to the typical laboratory oxidation where it is removed as H1. Note also that the hydride ion adds to the CACOCAN1 part of NAD1in a conjugate nucleophilic addition reaction, much as water might add to the CACOCAO part of an a,b-unsaturated ketone (Section 1.5).
1.9 Oxidations and Reductions 35
Nicotinamide Adenine C O NAD1 NADH (NADPH) NADH (NADP1) NAD1 C H O H B: N CONH2 CONH2 + + B H Alcohol Carbonyl compound N H H N N N N O2 O2 O OH OH O OH HO N C O O C N H H HO OH O OH OH O N N N N 1 1 1 NH2 NH2 NH2 NH2 O O P O P O CH2 O CH2 (OPO322) (OPO322) O2 O2 O O P O P O CH2 O CH2
Expulsion of hydride ion is not often seen in laboratory chemistry because, as noted at the beginning of Section 1.5, H2is a poor leaving group. One analo-gous process that does occur in laboratory chemistry, however, is the Cannizzaro reaction, which involves the disproportionation of an aromatic aldehyde on treat-ment with base. Benzaldehyde, for example, is converted to a mixture of benzyl alcohol and benzoic acid by reaction with NaOH. Hydroxide ion first adds to the aldehyde carbonyl group to give an alkoxide intermediate, which transfers a hydride ion to a second molecule of aldehyde. The first aldehyde is thereby oxi-dized, and the second aldehyde is reduced.
Biological reductions are the reverse of oxidations. As noted in Section 1.5, NADH transfers a hydride ion to the carbonyl group in a nucleophilic addition reaction, and the alkoxide intermediate is protonated.
H—A + .. C O NADH NAD1 + C H HO H H C O N N C O NH2 NH2 1 Cannizzaro reaction + HO....: C H O C OH O C H O C H HO C O2 H H 2 O: :..2
Problems
1.1 Which of the following substances can behave as either an acid or a base, depend-ing on the circumstances?
(a) CH3SH (b) Ca21 (c) NH3 (d)CH3SCH3
(e) NH41 (f) H2CACH2 (g) CH3CO22 (h)1H3NCH2CO22
1.2 Which CAC bond do you think is more nucleophilic, that in 1-butene or that in 3-buten-2-one? Explain.
1.3 Rank the following compounds in order of increasing acidity:
(a) Acetone, pKa519.3 (b) Phenol, pKa59.9
(c) Methanethiol, pKa510.3 (d) Formic acid, Ka51.99 31024
(e) Ethyl acetoacetate, Ka52.51 310211
1.4 Rank the following compounds in order of increasing basicity:
(a) Aniline, pKa(BH1)54.63 (b) Pyrrole, pKa(BH1)50.4
(c) Dimethylamine, pKa(BH1)510.5
(d) Ammonia, Ka(BH1)55.5 310210
1.5 Protonation of acetic acid by H2SO4might occur on either of two oxygen atoms. Draw resonance structures of both possible products, and explain why protonation occurs preferentially on the double-bond oxygen.
1.6 Protonation of a guanidino compound occurs on the double-bond nitrogen rather than on either of the single-bond nitrogens. Draw resonance structures of the three possible protonation products, and explain the observed result.
A guanidino compound C NH NH R .. .. .. H2N C O O H .. .. .. .. Acetic acid F H3C H2SO4 3-Buten-2-one 1-Butene F CH3CH2CH CH2 O CH3CCH CH2 Problems 37
1.7 Predict the product(s) of the following biological reactions by interpreting the flow of electrons as indicated by the curved arrows:
1.8 Complete the following biological mechanisms by adding curved arrows to indi-cate electron flow:
(b) B N H H CH3 CH3 OH N (a) .. .. .. .. N S R R9 C O O HO (c) O OPP O H3C H3C H3C 1 1 2 .. .. ..2 22O 3POCH2 OPO322 CO22 + + (a) O B O O (b) + R CH CH2 COCH3 O H : B : : R CH 2CH2 COCH3 BH1 + BH1 + CoAS2 O R C SCoA O O2 CH2 C R9 R0 R C CHR0 C R9 SCoA O O R C CHR0 C R9 O
1.9 Propose a mechanism for the following step in the b-oxidation pathway for degra-dation of fatty acids. HSCoA is the abbreviation for coenzyme A, a thiol.
1.10 The conversion of fructose 1,6-bisphosphate to glyceraldehyde 3-phosphate plus dihydroxyacetone phosphate is a step in the glycolysis pathway for degrading carbohydrates. Propose a mechanism.
1.11 Propose a mechanism for the conversion of 3-phosphoglycerate to phosphoenol-pyruvate (PEP), a step in the glycolysis pathway.
Phosphoenolpyruvate 2-Phosphoglycerate OPO322 HOCH2CHCO22 OPO322 H2C CCO22 Dihydroxyacetone phosphate Glyceraldehyde 3-phosphate Fructose 1,6-bisphosphate + CHO C H C O C O C HO H C H OH C O H H CH2OPO322 CH2OPO322 CH2OPO322 CH2OPO322 CH2OH OH + O O RCH2CH2CCH2CSCoA O RCH2CH2CSCoA O CH3CSCoA HSCoA Problems 39 (c) H—A H B : + BH1 H3C H3C H3C H3C H3C CO2CH3 CO2CH3 CO2CH3 CO2CH3 CO2CH3 CH3 CH3 CH3 CH3 CH3 1 1 1
1.12 The biological conversion of an aldehyde to a thioester occurs in two steps: (1) nucleophilic addition of a thiol to give a hemithioacetal, and (2) oxidation of the hemithioacetal by NAD1. Show the structure of the intermediate hemithioacetal, and propose mechanisms for both steps.
1.13 The loss of CO2(decarboxylation) from a b-keto acid happens frequently in bio-logical chemistry and takes place by a mechanism that is closely related to a retro-aldol reaction. Propose a mechanism for the following reaction that occurs in the citric acid cycle.
1.14 One of the biological pathways by which an amine is converted to a ketone involves two steps: (1) oxidation of the amine by NAD1to give an imine, and (2) hydrolysis of the imine to give a ketone plus ammonia. Glutamate, for instance, is converted by this process into a-ketoglutarate. Show the structure of the imine intermediate, and propose mechanisms for both steps.
1.15 The following reaction is part of the sequence by which pyruvate is converted to acetyl CoA. Propose a mechanism, and tell what kind of reaction is occurring.
b a a-Ketoglutarate O O O2 O Ab-keto acid + H1 2O 2C 2O2C CH2CO22 CO22 + CO2 Hemithioacetal F RCH2CH RCH2CSR9 R9SH NAD1 O O Imine Glutamate O a-Ketoglutarate 2O 2C 2O 2C CO22 CO22 + NH3 1NH 3 H2O NAD1 Lipoamide + N S R R9 OH .. S S R0 N S R R9 S HO SH R0 CH3 CH3 H3C H3C + H1 1
1.16 The amino acid methionine is formed by a methylation reaction of homocysteine with N-methyltetrahydrofolate. The stereochemistry of the reaction has been probed by carrying out the transformation using a donor with a “chiral methyl group” in which deuterium (D) and tritium (T) isotopes of hydrogen are present. Does the methylation reaction occur with inversion or retention of configuration? What mechanistic inferences can you draw?
Tetrahydrofolate N-Methyltetrahydrofolate Methionine synthase + Methionine N N N H O H NHAr C H T D Homocysteine N H C H T D HS CO22 CO22 H3N H3N H2N H H S 1 1 Problems 41