3537
Introduction
Bones are formed by one of two types of ossification processes, intramembranous or endochondral bone formation. The neurocranium is composed of membranous bones that are formed by direct ossification, and the sutures between them comprise apposing osteogenic fronts separated by mesenchymal cells (Opperman, 2000). This developmental process differs from endochondral bone formation at the base of the skull and long bones because the latter has an intermediate cartilage anlagen. In humans and mouse, these calvarial sutures appear at approximately 18 weeks or embryonic day 15 (E15) of gestation, respectively, and remain open at birth to allow continued brain growth. In mice, the two major midline sutures are the interfrontal (corresponding to metopic in humans) between the frontal bones and the sagittal
between the parietal bones. Sutures transverse to the midline include the coronal sutures between the frontal and parietal bones (see Fig. S1 in the supplementary material).
The midline interfrontal suture is neural crest derived, whereas, at the midline sagittal suture, there are neural crest cells present between the mesodermally derived parietal bones. The coronal suture lies at the interface of the neural crest-derived frontal bones and the mesoderm of the parietal bones (Jiang et al., 2002). At these sutures, a proportion of these cells are recruited to differentiate into osteoblasts and make new bone (Cohen and Kreiborg, 1993; Opperman, 2000). A cartilaginous layer underlies the lower part of the parietal bone and coronal sutures proximate to the endochondral bones at the base of the skull (Iseki et al., 1999). By contrast, there is no compelling evidence that cartilage is normally formed at the Apert syndrome is an autosomal dominant disorder
characterized by malformations of the skull, limbs and viscera. Two-thirds of affected individuals have a S252W mutation in fibroblast growth factor receptor 2 (FGFR2). To study the pathogenesis of this condition, we generated a knock-in mouse model with this mutation. The Fgfr2+/S252W mutant mice have abnormalities of the skeleton, as well as of other organs including the brain, thymus, lungs, heart and intestines. In the mutant neurocranium, we found a midline sutural defect and craniosynostosis with abnormal osteoblastic proliferation and differentiation. We noted ectopic cartilage at the midline sagittal suture, and
cartilage abnormalities in the basicranium, nasal turbinates and trachea. In addition, from the mutant long bones, in vitro cell cultures grown in osteogenic medium revealed chondrocytes, which were absent in the controls. Our results suggest that altered cartilage and bone development play a significant role in the pathogenesis of the Apert syndrome phenotype.
Key words: Apert syndrome, Fibroblast growth factor receptor 2, Mouse, Cartilage, Bone, Mesenchyme, Neural crest, Skull, Suture, Craniosynostosis
Summary
Abnormalities in cartilage and bone development in the Apert
syndrome FGFR2
+/S252Wmouse
Yingli Wang1,4,*, Ran Xiao1,4,*, Fan Yang5, Baktiar O. Karim2, Anthony J. Iacovelli1,4, Juanliang Cai1,4, Charles P. Lerner6, Joan T. Richtsmeier4,7, Jen M. Leszl7, Cheryl A. Hill7, Kai Yu8, David M. Ornitz8, Jennifer Elisseeff5, David L. Huso2and Ethylin Wang Jabs1,3,4,†
1Institute of Genetic Medicine, Department of Pediatrics, The Johns Hopkins University School of Medicine, 733 North Broadway,
Baltimore, MD 21205, USA
2Department of Comparative Medicine, The Johns Hopkins University School of Medicine, 733 North Broadway, Baltimore,
MD 21205, USA
3Department of Medicine and Plastic Surgery, The Johns Hopkins University School of Medicine, 733 North Broadway, Baltimore,
MD 21205, USA
4Center for Craniofacial Development and Disorders, The Johns Hopkins University School of Medicine, 733 North Broadway,
Baltimore, MD 21205, USA
5Department of Biomedical Engineering, The Johns Hopkins University, 3400 N. Charles Street, Clark 106, Baltimore, MD 21218,
USA
6Departments of Cell Biology/Microinjection and Microchemistry, The Jackson Laboratory, 600 Main Street, Bar Harbor,
ME 04609, USA
7Department of Anthropology, The Pennsylvania State University, 409 Carpenter Building, University Park, PA 16802, USA 8Department of Molecular Biology and Pharmacology, Washington University Medical School, 660 S. Euclid Avenue, St Louis,
MO 63110, USA
*These authors contributed equally to this work
†Author for correspondence (e-mail: ejabs1@jhem.jhmi.edu)
Accepted 13 May 2005
Development 132, 3537-3548
Published by The Company of Biologists 2005 doi:10.1242/dev.01914
Research article
Development and diseaseDe
superior part of the parietal bone and midline sutures of the membranous neurocranium.
In humans one of the most severe conditions involving abnormal sutural development and skull growth is Apert syndrome (Cohen, 2000). In infancy, a patent midline defect of the metopic and sagittal sutures, and synostosis (premature fusion) of the coronal suture, are present. During childhood, the sagittal and lambdoid sutures become synostosed (Kreiborg and Cohen, 1990). Other abnormalities of the skeleton include severe syndactyly, and internal organs are also affected, but less frequently (Table 1).
Remarkably, Apert syndrome is caused by a limited number of mutations. Ninety-nine percent of reported cases have one of two missense mutations in adjacent amino acids, Ser252Trp and Pro253Arg, of fibroblast growth factor receptor 2 (FGFR2) (Park et al., 1995; Wilkie et al., 1995). The first mutation, S252W, is more common, occurring in 67% of patients, and has been proposed to be associated with more severe craniofacial anomalies, whereas the second mutation, P253R, may be associated with more severe syndactyly (Slaney et al., 1996; von Gernet et al., 2000). Both mutations affect the highly conserved linker region between the immunoglobulin-like II and III domains and result in increased affinity and altered specificity of fibroblast growth factor (FGF) ligand binding (Ibrahimi et al., 2001; Yu et al., 2000).
Normally, the ligand-binding characteristics of FGFR2 vary depending on its isoform. Alternative splicing of exons IIIb and IIIc encoding the immunoglobulin-like III domain results in spliceforms that are expressed predominantly by epithelial or mesenchymal cells, respectively (Miki et al., 1992; Orr-Urtreger et al., 1993). Distinct ligands bind to the IIIb and IIIc isoforms. Ligands FGF7 and FGF10 activate FGFR2 IIIb, whereas FGFs 2, 4, 6, 8 and 9 activate FGFR2 IIIc. An Alu insertion flanking exon IIIc in an Apert syndrome patient was found to affect splicing and cause the ectopic expression of FGFR2 IIIb in fibroblasts from the patient (Oldridge et al., 1999).
FGFR2, as well as the other three FGFR tyrosine kinases and their 22 FGF ligands, is known to play a crucial role in the control of cell migration, proliferation, differentiation and survival, by activating two primary pathways, the mitogen-activating protein kinase pathway (Kouhara et al., 1997; Ornitz and Itoh, 2001) and protein kinase C pathway (Debiais et al., 2001; Lemonnier et al., 2001). Signaling through FGFR2 regulates stem cell proliferation, affecting different lineages such as osteoblasts and chondroblasts (De Moerlooze et al., 2000; Eswarakumar et al., 2002; Iseki et al., 1999; Ornitz and Marie, 2002).
Chen et al. created a transgenic mouse with an Fgfr2+/S250W
mutation (Chen et al., 2003). (Although the correct conserved serine at position 252 was targeted, it was incorrectly designated as residue 250 – Chu-Xia Deng, personal communication.) Their mice were found to have several features similar to Apert syndrome, including a small body size, brachycephaly, midface hypoplasia, short presphenoid bone, wide-spaced eyes, and malocculsion. The long bones revealed few abnormalities. The heights of the growth plates correlated with the smaller size of the mutants, and the column of proliferating chondrocytes was slightly short in older mice (postnatal day 10, P10). Most importantly, Chen et al. performed detailed studies of the transverse coronal sutures where they found premature closure, decreased bone formation, and increased apoptosis, but no obvious change in cell proliferation and differentiation. Thus, they suggest that dysregulated apoptosis plays an important role in the pathogenesis of Apert syndrome-related phenotypes.
In this study, we explored the pathogenesis of other Apert syndrome malformations, including the midline sutural abnormalities and those of the internal organs, by introducing the Fgfr2 S252W mutation into the mouse genome. Our resulting heterozygotes have phenotypic features consistent with the human Apert syndrome. This mutation alters proliferation and differentiation of osteoblasts at the midline calvarial sutures. It is also responsible for ectopic cartilage formation in the neurocranium and cartilaginous abnormalities in several organs including the long bones. These findings highlight the delicate balance of migration, proliferation and differentiation that orchestrate individual suture and bone development (Marie et al., 2002).
Materials and methods
Generation of targeting construct and mutant mice
We used a 6.28 kb genomic DNA fragment containing exons IIIa, IIIb and IIIc (exons 7, 8 and 9) of the murine Fgfr2 gene cloned into pBluescript SK– (Stratagene) to make a construct (Fig. 1A). A 755_756CA>GG substitution, resulting in a Ser252Trp mutation at the residue homologous to human FGFR2 amino acid 252, was introduced into exon IIIa using site-directed mutagenesis. We inserted a thymidine kinase (TK) gene cassette at the 5′XhoI site upstream of the targeting DNA fragment and a neo cassette with flanking loxP recognition sites into intron IIIa, oriented in an opposite transcriptional direction to the target Fgfr2. The final targeting vector of 13.6 kb was confirmed by sequencing, linearized by NotI digestion, and introduced into R1 ES cells by electroporation (carried out by The Jackson Laboratory).
[image:2.612.41.293.507.643.2]We identified two clones with the targeted mutant allele, and germline chimeric mice were generated from each clone (Fig. 1B). Table 1. Phenotypic comparison between Apert syndrome
patients and mice
Affected organ Humans* (%) Mice (%)
Skull 100 (68/68) 100 (10/10)
Craniosynostosis 100 (68/68) 67 (6/9)‡
Midline suture 100†(N/A) 100 (10/10)
Palate 58 (39/67) 100 (8/8)
Central nervous system 21 (10/48) 20 (2/10)§
Trachea Eight cases† 8 (1/13)§
Thymus No reported cases 50 (5/10)
Lungs 1.5†(2/136) 100 (13/13)
Cardiovascular system 16 (11/67) 64 (7/11) Gastrointestinal system 1.5†(2/136) 50 (5/10) Genitourinary system 8†(10/123) 0 (0/10)
Syndactyly 100 (68/68) 0 (0/13)
Long bones¶ 95†(36/68) 90 (9/10)
*Frequency in humans based on patients with a FGFR2+/S252W mutation (Cohen and Kreiborg, 1993; Park et al., 1995; Slaney et al., 1996).
†Based on patients with either a +/S252W or a +/P253R mutation (Cohen
and MacClean 2002).
‡Craniosynostosis based on abnormal histology in mice.
§Central nervous system and trachea not examined by serial sections in
mice.
¶Long bones refer to short humeri in humans and to abnormal histology in
mice.
N/A, not available.
De
Male chimeras were mated with C57BL/6J females to achieve germline transmission of the mutant allele. The presence of the neo cassette interfered with mutant allele expression. Therefore, F1 heterozygotes with neo (+/S252Wflox) were crossed with CMV or EIIA promoter Cre transgenic C57BL/6J mice (CMV-Cre, Transgenic Core Facility of the Johns Hopkins University School of Medicine; EIIA-Cre, The Jackson Laboratory) to remove the neocassette. Care and use of mice for this study were in compliance with the relevant animal welfare guidelines approved by the Johns Hopkins University Animal Care and Use Committee.
Mutant allele genotyping and RT-PCR expression analysis Genotypes of tail DNA from the resulting progeny were determined by PCR analysis using primers from within Fgfr2intron 6 (forward primer F, 5′-CTCGGAGTGTAACGTGTTC-3′), intron IIIa (reverse primer R, 5′-CAACAGGAAATCAAAGACC-3′) and the neo cassette (forward primer Fn, 5′-GATGTTTCGCTTGGTGGTC-3′) (Fig. 1A,C), and their identity was confirmed by DNA sequencing. We identified Fgfr2mutant allele expression in total RNA isolated from various organs and tissues from Fgfr2+/S252W mice by using the RNAqueousTM-4PCR Kit (Ambion) and the Omniscript RT Kit (QIAGEN) (Fig. 1D). RT-PCR amplification was performed using primers corresponding to the Fgfr2-coding sequence of exons 5 and 10 (forward primer, 5′-CAACACCGAGAAGATGGAG-3′and reverse primer, 5′-CCATGCAGGCGATTAAGAAG-3′). Products were digested with SfiI to distinguish the mutant from the wild-type allele.
Image analyses of the skull from the Fgfr2+/S252Wflox/S252W mouse
Adult mice were euthanized by halothane inhalation and X-rays were performed. After removing the skin, mice were decapitated and the heads were fixed in 4% paraformaldehyde. Micro-computed tomography scans were acquired by the Center for Quantitative Imaging at the Pennsylvania State University. Using eTDIPS software (http://www.cc.nih.gov/cip/software/etdips/), 3D reconstructions were made using the surface reconstruction module of eTDIPS, and 24 biological landmarks were located twice on each skull and mandible. The means of the three-dimensional coordinates of these landmarks from each mouse were used to analyze morphological differences between mice. A quantitative comparison of the linear distances estimated among the 24 landmarks using Euclidean Distance Matrix Analysis, or EDMA (http://oshima.anthro.psu.edu), provided precise measures of localized differences between mutant and control littermates.
Bone and cartilage staining, and measurements of skeleton
Skeletal staining with Alizarin Red S and Alcian Blue was performed (McLeod, 1980). Skull measurements, including skull and nose lengths, skull height and width, mandibular and maxillary lengths, and inner canthal distance (Richtsmeier et al., 2000), and limb measurements, including the length of the humerus, radius, ulna, femur, tibia and fibula, were taken with digital calipers (Fisher Scientific).
Histological analysis and special staining of the skeleton Whole heads of embryos at E14.5 to P1 were dissected. Histological sections (5 μm) were prepared from selected tissues that had been fixed in 4% paraformaldehyde and embedded in paraffin. Sections were stained with Hematoxylin and Eosin (HE) for histology. Alkaline phosphatase (ALP, specific for osteoblasts) and tartrate resistant acid phosphatase (TRAP, specific for osteoclasts) in skull sutures and growth plates were stained using the TRACP and ALP double-stain kit (TaKaRa Bio).
Immunohistochemical and TUNEL assays of skeleton Skull sutures, sectioned as above, were deparaffinized and hydrated through a xylene and graded alcohol series. The immunohistochemical
assays for Ki67 (Novocastra) and collagen II Ab-2 (Lab Vision) were performed using the VECTOR M.O.M Immunodetection Kit (Vector Laboratories). To assess cell proliferation, the slides were incubated with anti-Ki67 antibody, and visualized with biotinylated anti-mouse IgG (Vector) followed by the peroxidase reaction (brown color for Ki67-positive nuclei). Cell proliferation was analyzed by counting the number of Ki67-positive cells in more than 100 cells from a defined area, including and between the osteogenic fronts in three serial sections from four mutant and four control littermates for each developmental stage. The counts for Ki67-positive cells in mutant and wild-type embryos were compared by the t-test. To assess apoptosis, the TUNEL assay was performed, using the In Situ Cell Death Detection Kit, POD (Roche Applied Science) for detection of apoptotic cell death by light microscopy.
In situ hybridization
In situ hybridization was performed on sections as described by Wilkinson (Wilkinson, 1992) with modifications. The mouse osteonectin (ON), osteopontin (OP) and Sox9 cDNA fragments were each cloned into the pCR®II-TOPO® Vector. The plasmids were linearized, and antisense and sense single-stranded RNA probes were generated with the T7 and SP6 RNA polymerase.
In vitro culture
Cells were obtained from limbs of newborn Fgfr2+/S252Wmutant and wild-type mice. The long bones were dissected and connective tissue was removed. The bone samples from the middle third of the shaft were washed and centrifuged with phosphate-buffered saline (PBS, GIBCO). The samples were then digested in 1 mg/ml sterile collagenase D (Boehringer Mannheim) solution at 37°C for 2 hours, centrifuged and washed with PBS before resuspension in standard culture medium [Dulbecco’s modified Eagle’s medium (DMEM; GIBCO), 10% fetal bovine serum, 100 unit/ml penicillin and 100 μg/ml streptomycin]. Cells were plated onto a flask and medium changes occurred first after 3 days to allow cell attachment and then three times a week until confluency. Expanded cells were then placed in 3D culture (Elisseeff et al., 2005) and incubated in standard osteogenic medium for 3 weeks to evaluate differentiation. Osteogenic medium consisted of high-glucose DMEM, 100 nM dexamethasone (Sigma-Aldrich Co.), 50 µg/ml ascorbic acid 2-phosphate, 10 mM β -glycerophosphate, 10% fetal bovine serum, 100 unit/ml penicillin and 100 μg/ml streptomycin. Medium was changed every 2-3 days. At the end of 3 weeks in culture, wet and dry weights from all constructs for normalization of extracellular matrix content were obtained after 48 hours of lyophilization. ALP and calcium quantification, Von Kossa staining for mineralization (Pittenger et al., 1999), and immunohistochemical analyses for collagen type I and II (Research Diagnostics, rabbit polyclonal antibodies), were performed. For quantitative ALP assay, tissue cultures or constructs were homogenized in 0.75 M 2-amino-2-methyl propanol (Sigma; pH 10.3) solution and the supernatants were collected for ALP assay using Sigma ALP Determination Kits (Sigma Diagnostics 245), following the manufacturer’s protocol. For quantitative calcium measurement, lyophilized tissue constructs were homogenized in 0.5 M HCl and vigorously vortexed for 16 hours at 4°C. The supernatant was collected for calcium assay by following the manufacturer’s protocol (Sigma Diagnostics 587). Three samples from each group were harvested for each assay.
Results
Generation of Fgfr2+/S252Wmutant mice
We designed a gene-targeting construct to knock-in the human Apert FGFR2point mutation, site specifically, generating the homologous S252W amino acid change in the mouse (Fig. 1).
De
With the neo cassette, the mutant allele (S252Wflox) was
hypomorphic because it was either not expressed or its expression was reduced as determined by RT-PCR (see Table S1 in the supplementary material). Upon mating these mice with Cre transgenic mice, the neo gene was removed and resulting offspring expressed the mutant S252W allele in multiple organs and tissues, including the skull, sagittal and coronal sutures, palate, mandible, limbs, brain, thymus, heart, lung, liver, kidney, stomach and intestine (Fig. 1D, expression in skull and lung is shown). Heterozygous Fgfr2+/S252Wmutant
mice originating from two independently derived ES cell clones were born at the expected frequency of 25% and had the same phenotypic features. At birth they were smaller, and their weight was 83% that of their wild-type littermates [+/S252W (n=20) versus +/+ (n=112): 1.225±0.091 g versus 1.485±0.180 g, P<0.001]. Neonatal lethality occurred uniformly within 24 to 36 hours after birth due to apparent respiratory failure with palatal abnormalities, lung atelectasis and evidence of aerophagia. Although most humans with Apert syndrome survive the early postnatal period, a subset has been described with early postnatal death, thickened nasal cartilage, palate shape abnormalities, complete tracheal sleeve, and lung abnormalities (Cohen, 2000).
Skeletal and other organ abnormalities in Fgfr2+/S252Wmutant mice
Autopsy with skeletal preparations of mutant mice revealed multiple malformations that resembled those of human Apert syndrome (Fig. 2). A total of 33 Fgfr2+/S252W mutant mice were
analyzed at P1 (Table 1). The skull length, skull height and upper jaw length were significantly reduced when compared with control littermates [+/S252W (n=4) versus +/+ (n=4): average skull length 9.415 mm versus 10.450 mm, P=0.006; skull height 6.135 mm versus 6.570 mm, P=0.028; upper jaw length 5.399 mm versus 6.200 mm, P=0.018]. Their skull width, nose length, inner canthal distance, and lower jaw measurements did not differ from those of controls (P=0.052 to 0.842).
[image:4.612.42.367.333.743.2]Other skeletal system abnormalities included increased cartilage of the basicranium, malformation of the palate, thickened nasal cartilage, fusion of joints separating the zygomatic arch bones, fusion of bones of the sternum, and complete cartilage sleeve of the trachea (Fig. 2). Although the mutant mice were smaller, the upper and lower limb lengths were proportional between mutant and control littermates (ratio of humerus/radius in +/S252W versus +/+: 0.836 versus 0.838, P=0.975; ratio of femur/tibia in +/S252W versus +/+:
Fig. 1. Generation of the targeting construct and Fgfr2+/S252Wmice. (A) Structure of a portion of the wild-type Fgfr2gene with exons IIIa, IIIb, IIIc and 10; the targeting construct with the TK cassette and neocassette with flanking loxPsequences; and the mutant allele produced by homologous recombination and with neodeletion mediated by Cre. The 755_756CA>GG, S252W mutation (*) was introduced into exon IIIa. Probes (–) and restriction enzymes (St, StyI; H, HindIII; S, SacI) used for Southern blot analysis, and PCR primers (F, Fn, R; arrowheads) used for genotyping are shown. (B) Identification of mutant and wild-type alleles in two
independent ES cell clones using Southern blot analysis with 5′and 3′probes. (C) Genotype of a litter from +/S252Wflox+/Cre by PCR of tail DNA. Lanes 1, 3 and 5: heterozygote with +/S252Wflox, showing wild-type allele (521 bp) and hypomorphic allele with neo (1.1 kb). Lanes 2 and 4: heterozygote mutant +/S252W, showing mutant allele after neodeletion (581 bp). Lane 7: hypomorphic mutant
+/S252Wflox/S252W. Lanes 6 and 8: wild type (+/+). (D) RT-PCR detection of mutant transcript in lung and skull tissues. Other tissues tested expressed the mutant allele (data not shown). The mutation introduces an additional SfiI restriction site. (E) Gross appearance of +/S252W and wild-type postnatal day (P)1 mice. Note the small body size of the mutant mouse. The difference of body weight between the mutant and control mice was significant.
De
0.616 versus 0.595, P=0.790). None of the mutants exhibited syndactyly.
Histopathological analysis of the brain revealed mild hydrocephalus with enlarged ventricles (Fig. 2). In the lungs, moderate and diffuse atelectasis with lack of alveolar expansion and the presence of proteinaceous fluid in the alveoli were observed. Multifocal and acute bronchiolectasis with mild dilation of the bronchioles were found. Within the thymus there was diffuse lymphoid necrosis and apoptosis of varying severity (data not shown). Cardiovascular abnormalities included dilation of the atria and great vessels at the base of the heart, and mild luminal ectasia was found in the stomach and the intestine, consistent with aerophagia (data not shown).
Skull abnormalities in the Fgfr2+/S252Wflox/S252W mutant mice
We analyzed the only two mutant mice with the hypomorphic S252Wfloxallele that survived to adulthood. In these mice, the
neocassette was not completely removed in all tissues (Fig. 1C, lane 7). The expression levels of both mutant Fgfr2 IIIb
[image:5.612.160.562.77.479.2]and IIIc alternative transcripts in hypomorphic mutant mice were generally reduced by 16% to 41%, when compared with those of the normal transcripts in multiple tissues examined, including lung, liver, kidney and stomach (see Table S1 in the supplementary material). At 10 months, these mutants had an abnormal head shape, with a body weight that was 29% (mutant 10.63 g versus +/+ 36.35 g) and a length that was 68% (nasal tip to base of tail, mutant 62.69 mm versus +/+ 92.27 mm) of those of their littermate controls (Fig. 3). Micro-CT of the adult skull revealed multiple abnormalities with an interfrontal sutural defect (Fig. 3C, arrow), dysmorphology of the nasal bones, and obliteration of the fronto-premaxillary, premaxillary-maxillary, and nasal-frontal sutures. The structures of the face and palate were 40-50% smaller in the mutant mouse than in the control mouse, while structures of the neurocranium and basicranium were reduced by 30-40%. Linear distances measuring the width of the skull were affected less than the length was, resulting in a very brachycephalic skull shape. The frontal process of the maxilla showed an abnormally large sinus and bowing of the medial wall and the Fig. 2.Gross bony structures
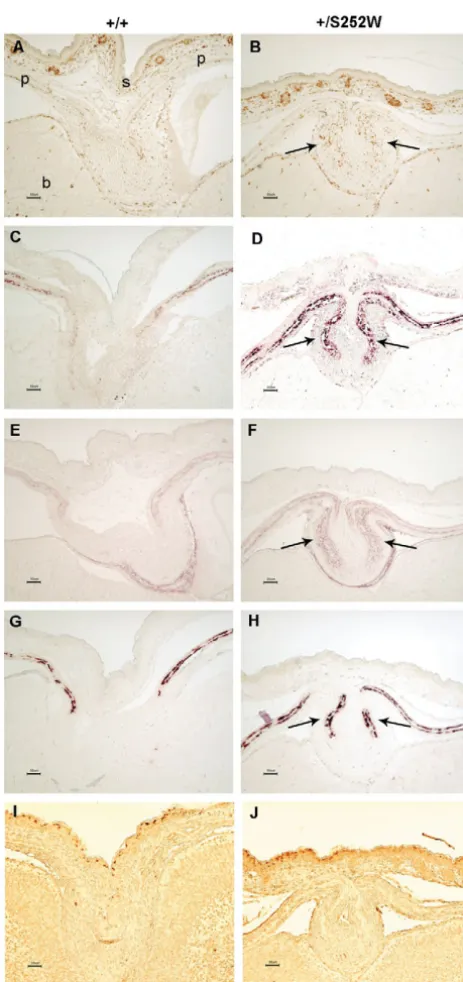
and histology of P1 Fgfr2+/S252Wmice.
(A,C,E,G,I,K,M,O,Q,S) Control littermates;
(B,D,F,H,J,L,N,P,R,T)
corresponding regions in mutant mice. (A-H) Alizarin Red S and Alcian Blue staining of (A-F) the skull and (G,H) the chest. (I-R) HE staining of the (I,J) brain, (K,L) palate, (M,N) trachea, (O,P) lung, and (Q,R) growth plates of long bone. (S,T) TRAP staining for growth plates. The mutant mice have: a wide interfrontal suture (B, arrows define the width of the suture); a shortened nasal snout (D, arrow); fusion of the joints between the zygomatic arch bones (F, arrows); a sternum with abnormal fusions and bifid xiphoid process (H, arrow); hydrocephalus with enlarged ventricles (J, arrows); a palate defect (L, arrow); a complete cartilage sleeve of the trachea with abnormal cartilage thickening (N); atelectasis of the lungs with alveolar proteinaceous fluid (P); chondrocyte hypertrophy and disorganization with irregular and thickened ossifying cartilaginous spicules in the growth plates of long bone (R); and decreased osteoclasts at the chondro-osseous junction (T; S, arrow shows normal osteoclasts
stained red). LV, lateral ventricle; 3V, third ventricle; TC, tracheal cartilage; P, zone of chondrocyte proliferation; H, zone of hypertrophy; M, zone of mineralization; O, zone of ossification at the chondro-osseous junction. Scale bars: A-H, 1 mm; I-T, 50 μm.
De
zygomatic process of the maxilla was more bowed laterally. The interparietal bone was compressed superiorly at the lambdoid suture whereas the parietal bones showed an obvious superior bulge. The inner surface of the calvarium showed ‘thumb printing’ or fine indentations presumed to be secondary to increased intracranial pressure. The mandible was very small with a dysmorphic angular process.
Sutural dysmorphology in Fgfr2+/S252Wmutant mice Sutures were examined microscopically to further characterize the abnormalities observed in the mutant skulls during early mouse development. Individual sutures exhibited different histological findings. The midline sagittal suture was similar in both mutant and control littermates at E16.5. In E18.5 mutants, the space between the two osteogenic fronts of the sagittal suture was reduced with increased cellularity. In P1 mutants, the osteogenic fronts were very proximate, and osteoid had begun to be deposited between them prior to synostosis of the parietal bones; by contrast, in the control littermates, the osteogenic fronts were widely separated (Fig. 4A-F).
The midline interfrontal suture showed an obvious gap between the osteogenic fronts of the frontal bones in the mutant mice when compared with control littermates (Fig. 4G,H). The widely separated osteogenic fronts correlate with the patent midline defect observed macroscopically in skeletal preparations (Fig. 2B) and CT scans of mutant mice (Fig. 3C, see arrow). These results in mice are consistent with the midline sutural defect found in all Apert syndrome infants (Kreiborg and Cohen, 1990).
The transverse coronal (Fig. 4I,J) and lambdoid sutures (Fig. 4K,L) in P1 mutants showed synostosis with osteoid deposition. Our results are consistent with the coronal synostosis found in P1 to P18 Apert syndrome mutant mice reported by Chen et al. (Chen et al., 2003) and on CT scans of Apert syndrome patients (Kreiborg and Cohen, 1990).
Abnormal osteoblastic proliferation and
differentiation at the midline sutures in mutant mice The effects of the Fgfr2 +/S252W mutation on proliferation, differentiation and apoptosis were investigated at the sagittal suture from E16.5 to P1. Cell proliferation was investigated by counting Ki67-positive cells from a designated area in the sagittal sutures using serial sections. At E16.5, no difference in the number or distribution of Ki67-positive cells was apparent between mutants and controls (data not shown). At E18.5 and P1, there were twice as many proliferating Ki67 cells at the sagittal suture in mutants (E18.5 +/S252W versus +/+: 98.0±5.0 positive cells versus 45.5±10.6 positive cells,
[image:6.612.38.420.75.401.2]P<0.023; P1 94.5±10.6 versus 26.5±0.7, P<0.018; Fig. 5A,B). These mutant Ki67-positive cells were abnormally distributed in the sutural space between the osteogenic fronts, as well as being localized at the fronts where they are present in controls. Osteoblast differentiation was assessed using osteonectin (ON), osteopontin (OP) and alkaline phosphatase (ALP) markers. At E16.5, no differences were apparent in the expression of these markers between mutants and controls (data not shown). At E18.5, ON, OP and ALP were expressed in the developing bone and in cells extending from the margin
Fig. 3.Skeletal features of hypomorphic Fgfr2+/S252Wflox/S252W mice. (A) The gross appearance shows the smaller body size of the mutant mouse at 8 weeks of age. (B) X-ray of the whole body at 8 weeks of age shows short nasal bones and maxillary bones with overriding lower incisors. (C) Micro-CT of the skull of a 10-month-old Fgfr2+/S252Wflox/S252W mouse (right) shows extreme brachycephaly, diminutive and curved nasal bones, extreme reduction anteroposteriorly of the frontal bones, obliterated fronto-nasal, fronto-premaxillary and maxillary-premaxillary sutures, and a defect of the interfrontal suture in the skull (arrow).
De
of the bone plates into the mid-sutural area in mutants (Fig. 5C-H). These results suggest abnormal osteoblastic differentiation, because this latter area normally contains mesenchymal or osteoprogenitor cells, but no preosteoblasts. Apoptosis at the sagittal suture was studied by TUNEL staining. There was no apparent difference in apoptosis
between mutants and controls at E16.5, E18.5 and P1 (Fig. 5I,J; E18.5 is shown).
We also examined proliferation, differentiation and apoptosis at the midline interfrontal suture. In mutants, there was a significant increase in the number of Ki67-positive cells between the osteogenic fronts (E18.5 +/S252W versus +/+: 181.0±4.2 positive cells versus 111.0±8.5 positive cells,
P<0.01). The mutant Ki67 cells had a pronounced alteration in their distribution. Rather than being enriched at the osteogenic fronts, Ki67-positive cells in the mutant extended from the widely separated margins of the osteoid plates almost into the mid-sutural space (Fig. 6C,D). In situ hybridization with ON and OP revealed an obvious lack of differentiating osteoblasts between the osteogenic fronts in mutants (Fig. 6E-H). This difference could be detected at E16.5 and became more obvious at E18.5. Similar to our results at the sagittal suture, there was no obvious difference in apoptosis at the interfrontal suture of mutants and controls from E16.5 to E18.5 (data not shown).
Abnormal cartilage at the midline sagittal suture in mutant mice
In Fgfr2+/S252Wmice, there was ectopic cartilage at the sagittal
suture (n=20 mutants, n=26 controls; Fig. 7). Normally, with intramembranous bone formation at the sagittal suture, there is no intermediate cartilaginous stage. At E16.5, cartilage was detected consistently throughout most of the entire length of the sagittal suture of mutants. At E18.5, cartilage was present along more than half of the length of the sagittal suture (0.45±0.11 mm of 0.71± 0.05 mm, as determined by 5 μm serial sections), beginning at the junction between the parietal and interparietal bones (see Fig. S1 in the supplementary material). At P1, the cartilage resembled that at E18.5 in the sagittal suture of mutants. Two chondrogenic markers, Sox9 and collagen type II, were expressed in these cells (Fig. 7E-L). No chondrocytes or cartilage was detected histologically, and no Sox9 expression was present in mutant sagittal sutures at earlier stages of E14.5 or E15.5 (data not shown) or in controls from E14.5 to E18.5. At the later P1 stage, cartilage was detected near the junction of the parietal and interparietal bones at the sagittal suture in only one out of the 26 controls.
[image:7.612.54.285.152.735.2]Abnormal cartilage in the limb of mutant mice The long bones were analyzed by histopathology. There was disorganization of the growth plates with subtle irregularity of the hypertrophic zone and more prominent cartilage
Fig. 4. Histological analysis of the calvarial sutures in Fgfr2+/S252W mice. (A,C,E,G,I,K) Control littermates; (B,D,F,H,J,L)
corresponding regions in mutant mice. (A-F) HE staining shows the development of the sagittal suture from E16.5 to P1. (A,B) No obvious difference of the sagittal sutures between the mutant and control at E16.5. (C,D) More proximate osteogenic fronts are evident at the mutant sagittal suture at E18.5. (E,F) Osteoid deposition is seen across the mutant sagittal suture at P1. (G-L) HE staining shows: a wide gap between the osteogenic fronts at the mutant interfrontal suture at P1 (G,H); presynostosis/synostosis with osteoid deposition at the mutant coronal suture at P1 (I,J); and
presynostosis/synostosis at the mutant lambdoid suture at P1 (K,L). Arrows, osteogenic fronts; arrowheads, presynostosis/synostosis with osteoid formation; s, skin; p, parietal bone; b, brain; f, frontal bone; ip, interparietal bone. Scale bars: A-H,K,L, 50 μm; I,J, 25 μm.
De
mineralization at P1. Markedly decreased osteoclasts at the chondro-osseous junction were observed by TRAP staining (Fig. 2Q-T). Samples were isolated from the long bones of mutant mice to obtain further evidence of abnormal cartilage
[image:8.612.49.281.125.616.2]formation. Before culture, both mutant and wild-type cells represented a heterogeneous population. They were positive for bone-related proteins (ALP, collagen type I) and cartilage-related protein collagen type II (see Fig. S2 in the supplementary material). Then these cells were cultured in established osteogenic medium. After three weeks in culture, both mutant and wild-type cells still generated bone-related proteins ALP and Col I. Von Kossa staining showed mineralization in the pericellular regions, which was also confirmed by quantitative calcium assays (+/S252W versus +/+: 4.25±0.38% versus 3.66±0.67% dry weight). Chondrocytes were detected by collagen type II staining of mutant cells cultured in 3D osteogenic conditions, but were absent in control cells (Fig. 8E,F). The DNA content did not differ between mutant and wild-type cultures assayed at 1 day and 21 days of growth, suggesting that increased proliferation may not be the explanation for the presence of mutant
Fig. 5. Proliferation, differentiation and apoptosis at the midline sagittal suture in Fgfr2+/S252Wmice at E18.5.
[image:8.612.320.553.275.630.2](A,B) Immunohistochemical staining of Ki67 shows increased numbers and abnormal distribution of positive cells indicating abnormal proliferation in mutants (arrows). (C-H) Expression of osteogenic markers shows increased or abnormal differentiation in mutants. (C,D) Osteonectin in situ hybridization (arrows). (E,F) ALP staining (arrows). (G,H) Osteopontin in situ hybridization (arrows). (I,J) TUNEL staining does not show an obvious difference in apoptosis between mutants and controls. (A,C,E,G,I) Control littermates; (B,D,F,H,J) corresponding regions in mutant mice. s, skin; p, parietal bone; b, brain. Scale bars: A-J, 50 μm.
Fig. 6.Proliferation and differentiation at the midline interfrontal suture in Fgfr2+/S252Wmice at E18.5. (A,B) HE staining shows a gap in mutants. (C,D) Ki67 staining shows abnormal distribution of positive cells in the mid-sutural mesenchyme in mutants.
(E-H) Expression of osteogenic markers shows displaced, decreased or delayed differentiation in mutants. (E,F) Osteonectin in situ hybridization; (G,H) osteopontin in situ hybridization.
(A,C,E,G) Control littermates; (B,D,F,H) corresponding regions in mutant mice. Arrows, osteogenic fronts; s, skin; f, frontal bone; b, brain. Scale bars: A-H, 50 μm.
De
chondroctyes in culture (see Fig. S3 in the supplementary material). Perhaps, the osteogenic medium does not support the growth of normal chondrocytes and does not inhibit that of mutant cells.
Discussion
Apert syndrome mouse model
Using a Cre-mediated knock-in method, we have generated mice carrying the S252W mutation, which is the most common FGFR2 mutation associated with Apert syndrome in humans. The mutant mice serve as a model for Apert syndrome, as their phenotype is consistent with the human condition (Table 1).
Fgfr2+/S252W mice are neonatal lethal, small in size, and
demonstrate significant anomalies, including craniosynostosis and multiple visceral anomalies, as were evident by our histopathological studies. A high penetrance of affected organs in this mouse model indicates that signaling through Fgfr2 is crucial to the normal development of the skeleton, as well as affecting the brain, lungs, thymus, and other visceral organs. Survival of the hypomorphic, Fgfr2+/S252Wflox/S252W mutant mice makes them useful in studying the postnatal skull abnormalities related to this condition.
We confirmed the results of Chen et al. (Chen et al., 2003), including the presence of coronal synostosis in mutant mice. In addition, our analysis focused on the midline sagittal and interfrontal sutures, where a skull defect is present in all human Apert syndrome newborns. Although, severe syndactyly is a
key feature of Apert syndrome in humans, none of the mutant mice from either laboratory exhibited syndactyly by gross inspection and histopathology. The absence of this feature is consistent with the possibility that this mutation causes less severe digital abnormalities in humans when compared with the Apert FGFR2 P253R mutation (Slaney et al., 1996; von Gernet et al., 2000). Others speculate that there is no human phenotypic difference for these two mutations (Park et al., 1995). Thus, the absence of syndactyly in our mice may be a consequence of species-specific modifications or of alternative splicing affecting levels of expression of receptors (Oldridge et al., 1999).
Abnormal bone development at the midline calvarial sutures in Apert syndrome
Previously in the literature there has been few, if any, histopathological studies of the midline sutures, especially the interfrontal suture, in craniosynostosis mouse models. For the
Fgfr2+/S252W mutant, the interfrontal suture had increased proliferation, but abnormally localized, decreased or delayed differentiation of osteoprogenitor cells to preosteoblasts. Therefore, abnormal proliferation and differentiation of osteoblasts, not increased apoptosis, may underlie the metopic sutural defect in Apert syndrome.
[image:9.612.64.562.71.366.2]At the mutant sagittal suture, the number of proliferating cells and the expression of osteogenic markers were mislocalized and increased, suggesting that the observed synostosis results from increased proliferation and abnormal differentiation. In mutant Fig. 7. Abnormal cartilage at the midline sagittal suture in Fgfr2+/S252Wmice from E16.5 to E18.5. (A-D) HE staining, (E-H) Sox9 in situ hybridization, and (I-L) collagen type II immunohistochemical staining, showing the abnormal cartilage in mutants from E16.5 (left panels) to E18.5 (right panels). (A,E,I,C,G,K) Control littermates; (B,F,J,D,H,L) corresponding regions in mutant mice. Arrows, abnormal cartilage; s, skin; p, parietal bone; b, brain. Scale bars: A-L, 50 μm.
De
mice, the osteogenic markers are expressed abnormally in cells extending from the margin of bone plates to the mid-sutural area. It is possible that the Fgfr2 +/S252W mutation may cause abnormal differentiation at the developing sagittal sutures by altering the fate of mesenchymal cells.
Although there is increased and abnormal proliferation at both mutant midline sutures, we observed significant differences in their development. The abnormal differentiation at the sagittal suture leads to synostosis between the parietal bones, while at the interfrontal suture it results in the gap between the frontal bones. By contrast, in the mutant coronal suture, there is no increased proliferation or differentiation, but increased apoptosis that leads to synostosis (Chen et al., 2003). Our findings demonstrate that bone development in Apert syndrome can vary among specific sutures and even sutural synostoses can be due to either increased growth by proliferation and differentiation, or decreased growth with apoptosis across a suture.
Abnormal cartilage development in Apert syndrome We provide strong evidence that abnormal cartilage formation is involved in the pathogenesis of Apert syndrome. In humans,
although cartilage abnormalities have been reported in this condition, as well as in other craniosynostosis syndromes such as Crouzon and Pfeiffer (Cohen and Kreiborg, 1992; Cohen and Kreiborg, 1993; Kreiborg et al., 1993; Kreiborg et al., 1999), abnormal cartilage formation has not previously been recognized as being significant. In our mouse model, the Fgfr2 +/S252W mutation has a generalizable effect on the development of cartilage. Chondrogenic markers and increased cartilage were found in the mutant sagittal suture at embryonic stages when normally they are not present. These findings suggest that either there was a significant increase in cartilage proliferation at the junction of the parietal and interparietal bones, where it normally may be present, or that there is ectopic cartilage formation from progenitor cells. We also observed increased cartilage at the basicranium, nasal turbinates, trachea and long bones of our mutant mice. At the long bone growth plates, abnormal cartilage formation was detected histologically in vivo, and this was further supported by our in vitro cultures. These results indicate that the Fgfr2 +/S252W mutation significantly enhances the formation of cartilage in some organs, and we speculate that abnormal chondrogenesis may occur.
Interestingly, our results differ for the two midline sutures. At the sagittal suture, there is ectopic cartilage and synostosis, and at the interfrontal suture there is a defect where no cartilage was found. The distinct tissue origins of the sagittal and interfrontal sutures may reflect their mutant osteogenic potentials. The sagittal suture is formed from neural crest cells and mesoderm, whereas the interfrontal suture is neural crest derived (Jiang et al., 2002). The cranial neural crest cells can give rise to either cartilage or bone (Le Douarin et al., 1993; Noden, 1983; Selleck et al., 1993). In vitro studies have shown that mutant FGFRs can induce ectopic cartilage from premigratory neural crest cells early in development. Petiot et al. demonstrated that transfection of activating FGFR1 K656E or FGFR2 C278F mutations can induce cartilage differentiation when electroporated into quail premigratory neural crest cells (Petiot et al., 2002), but this effect is drastically reduced if transfection is carried out after the onset of neural crest migration. Therefore, the observed abnormal development of cartilage in Apert syndrome may be due to the mis-migration or mis-positioning of neural crest cells at an early embryonic stage.
Of note, there have been previous reports of abnormal cartilage formation in the sagittal suture in pathologic states. In mutant mice overexpressing Msx2, cartilage has been demonstrated to underlie the midline sagittal suture (Liu et al., 1999). Mice, exposed to retinoic acid at E10, develop cartilage in the parietal region concomitant with the decrease in the intramembranous ossification (Jiang et al., 2002), suggesting that neural crest or mesenchymal cell fate may be sensitive to Msx2 dosage. Rice et al. reported that cartilaginous rods are occasionally and transiently seen in the sutural mesenchyme around the time of birth (Rice et al., 2000), and this may be a tissue reaction to mechanical irritation.
[image:10.612.46.280.69.409.2]The complexity in the precise temporospatial definition of each developing suture in Apert syndrome is underscored by the variable effects of the Fgfr2 +/S252W mutation, differences in tissue origins, and potential genetic and environmental modifiers. These distinctions cannot be easily studied in human surgical or cultured cells because of the lack of specimens at Fig. 8.Chondrocytes in cultured cells from the limb of P1
Fgfr2+/S252Wmice. (A,B) Von Kossa staining at the pericellular regions and (C,D) collagen type I immunohistochemical staining are similar for both mutants and controls. (E,F) Collagen type II staining is positive for the mutants, but is absent in controls. (A,C,E) Control littermates; (B,D,F) mutants. Scale bars: A-F, 100 μm.
De
different developmental stages, sites, and controls. In fact, previous studies on human samples have differing results. Lomri et al. (Lomri et al., 1998) and Lemonnier et al. (Lemonnier et al., 2000) concluded that there was increased osteoblast differentiation of calvarial cells without making a distinction between sagittal, metopic and coronal synostosed sutures in Apert syndrome fetuses and infants. Mansukhani et al. (Mansukhani et al., 2000) noted an opposite effect of inhibition of differentiation, and increased apoptosis in immortalized human osteoblasts, not specifically derived from sutures, introduced with the FGFR2 S252W mutation.
Insights into the mechanism of the Fgfr2 +/S252W mutation
Mutant mice with loss of function or dominant-negative mutations in Fgfr2 showed abnormalities in some of the same organs that are affected in our Apert syndrome mouse model. Mice homozygous for null alleles of Fgfr2 die at E10.5 with multiple defects in organogenesis, and chimeras between this mutant and wild-type had abnormal epithelial-mesenchymal interactions, respiratory failure and intramembranous and endochondral bone formation (Arman et al., 1998; Arman et al., 1999; Xu et al., 1998). A mouse with a specific deletion and a dominant-negative loss of Fgfr2 IIIb isoform function resulted in craniofacial (wide cleft palate, reduced maxillary bone, absent otic capsule, rudimentary inner ear structures) and truncating limb anomalies, dysgenesis of several visceral organs (thymus, glandular stomach, pancreas, kidney), and agenesis of the tooth bud, salivary gland, thyroid, pituitary and lung (Celli et al., 1998; De Moerlooze et al., 2000).
Mice created by a specific deletion of Fgfr2 IIIc function associated with exon switching more closely resemble our Apert syndrome mice. Hemizygotes were affected with neonatal growth retardation and death, coronal synostosis, ocular proptosis with fused joints of the zygomatic arch bones, precocious sternal fusion, and abnormalities in secondary branching in several organs, such as the lungs, kidneys and lacrimal glands (Hajihosseini et al., 2001). With a splice-switch mechanism, Fgfr2 IIIb expression was increased in calvarial sutures and the zygomatic joints allowing cells, which usually express Fgfr2 IIIc, to respond to a broader set of ligands. Similarity between this mouse mutant and the Apert syndrome mouse suggests that the Fgfr2 +/S252W mutation is neomorphic as supported by the observed change in ligand-binding specificity (Yu et al., 2000), rather than being a ‘simple’ gain-of-function mutation (Anderson et al., 1998).
Mice created by conditional inactivation of Fgfr2 with Dermo1cre/+, specifically targeting Cre expression to disrupt signaling in the chondrocyte or osteocyte lineages, gave further insight into the potential mechanism of the Apert mutation (Yu et al., 2003). These mutant mice had a few features, such as a domed-shaped skull with a midline interfrontal sutural defect, that are similar to our Apert syndrome model. They also had a dwarfism phenotype with a shortened axial and appendicular skeleton that differed from our mice by a reduced hypertrophic chondrocyte zone, vertebral abnormalities with non-ossified gap in the dorsal midline of both cervical and thoracic vertebrae, absence of the spinous processes, and a lack of tarsal joints because of failure of cavitation of the cartilaginous anlage. They demonstrated that Fgfr2 is essential for the proliferation of osteoprogenitors and for the maintenance of
osteoblast anabolic function, but that it is not required for osteoblast differentiation. The phenotype of their mice is different from our Apert syndrome mice because the mechanism of the Fgfr2 +/S252W mutation is not inactivation. Analysis of our mouse model establishes a potential link between abnormal proliferation and differentiation, and possibly altered cell fate determination of progenitor cells with developmental abnormalities in Apert syndrome. Abnormal chondrogenesis may play a role in the pathogenesis of craniosynostosis syndromes. The Apert mouse model provides an in vivo system for future studies of determinants of cell fate for the chondrocyte or osteoblast lineages, which are important in the larger context of bone cell biology.
This work was supported by NIH grants DE11441 (E.W.J.), DE13078 (E.W.J. and J.T.R.), F33DE/HD05706 (J.T.R.), HD38384 (J.T.R.), HD24605 (J.T.R.), RR00171 (D.L.H.) and HD39952 (D.M.O.).
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/cgi/content/full/132/15/3537/DC1
References
Anderson, J., Burns, H. D., Enriquez-Harris, P., Wilkie, A. O. and Heath, J. K.(1998). Apert syndrome mutations in fibroblast growth factor receptor 2 exhibit increased affinity for FGF ligand. Hum. Mol. Genet. 7, 1475-1483.
Arman, E., Haffner-Krausz, R., Chen, Y., Heath, J. K. and Lonai, P.
(1998). Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development.Proc. Natl. Acad. Sci. USA95, 5082-5087.
Arman, E., Haffner-Krausz, R., Gorivodsky, M. and Lonai, P. (1999). Fgfr2 is required for limb outgrowth and lung-branching morphogenesis.
Proc. Natl. Acad. Sci. USA96, 11895-11899.
Celli, G., LaRochelle, W. J., Mackem, S., Sharp, R. and Merlino, G.(1998). Soluble dominant-negative receptor uncovers essential roles for fibroblast growth factors in multi-organ induction and patterning. EMBO J. 17, 1642-1655.
Chen, L., Li, D., Li, C., Engel, A. and Deng, C. X.(2003). A Ser250Trp substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone 33, 169-178.
Cohen, M. M., Jr(2000). Apert syndrome. In Craniosynostosis Diagnosis,
Evaluation, and Management(eds. M. M. Cohen, Jr and R. E. McLean), pp.
316-353. New York: Oxford University Press.
Cohen, M. M., Jr and Kreiborg, S. (1992). New indirect method for estimating the birth prevalence of the Apert syndrome. Int. J. Oral. Maxillofac. Surg. 21, 107-109.
Cohen, M. M., Jr and Kreiborg, S.(1993). Visceral anomalies in the Apert syndrome. Am. J. Med. Genet. 45, 758-760.
Debiais, F., Lemonnier, J., Hay, E., Delannoy, P., Caverzasio, J. and Marie, P. J. (2001). Fibroblast growth factor-2 (FGF-2) increases N-cadherin expression through protein kinase C and Src-kinase pathways in human calvaria osteoblasts. J. Cell Biochem. 81, 68-81.
De Moerlooze, L., Spencer-Dene, B., Revest, J., Hajihosseini, M., Rosewell, I. and Dickson, C. (2000). An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development127, 483-492.
Elisseeff, J., Kim, T. K., Ruffner, M. and Williams, C. G. (2005). Cellular photoencapsulation in hydrogels. In Culture of Cells for Tissue Engineering
(eds. I. Freshney and G. Vunjak-Novakovic). Hoboken, NJ: John Wiley and Sons (in press).
Eswarakumar, V. P., Monsonego-Ornan, E., Pines, M., Antonopoulou, I., Morriss-Kay, G. M. and Lonai, P.(2002). The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development129, 3783-3793.
Hajihosseini, M. K., Wilson, S., De Moerlooze, L. and Dickson, C.(2001). A splicing switch and gain-of-function mutation in Fgfr2-IIIc hemizygotes causes Apert/Pfeiffer-syndrome-like phenotypes. Proc. Natl. Acad. Sci. USA
98, 3855-3860.
Ibrahimi, O. A., Eliseenkova, A. V., Plotnikov, A. N., Yu, K., Ornitz, D. M.
De
and Mohammadi, M.(2001). Structural basis for fibroblast growth factor receptor 2 activation in Apert syndrome. Proc. Natl. Acad. Sci. USA98, 7182-7187.
Iseki, S., Wilkie, A. O. and Morriss-Kay, G. M.(1999). Fgfr1 and Fgfr2 have distinct differentiation- and proliferation-related roles in the developing mouse skull vault. Development126, 5611-5620.
Jiang, X., Iseki, S., Maxson, R. E., Sucov, H. M. and Morriss-Kay, G. M.
(2002). Tissue origins and interactions in the mammalian skull vault. Dev. Biol. 241, 106-116.
Kouhara, H., Hadari, Y. R., Spivak-Kroizman, T., Schilling, J., Bar-Sagi, D., Lax, I. and Schlessinger J.(1997). A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell89, 693-702.
Kreiborg, S. and Cohen, M. M., Jr(1990). Characteristics of the infant Apert skull and its subsequent development. J. Craniofac. Genet. Dev. Biol. 10, 399-410.
Kreiborg, S., Marsh, J. L., Cohen, M. M., Jr, Liversage, M., Pedersen, H., Skovby, F., Borgesen, S. E. and Vannier, M. W.(1993). Comparative three-dimensional analysis of CT-scans of the calvaria and cranial base in Apert and Crouzon syndromes. J. Craniomaxillofac. Surg. 21, 181-188.
Kreiborg, S., Aduss, H. and Cohen, M. M., Jr(1999). Cephalometric study of the Apert syndrome in adolescence and adulthood. J. Craniofac. Genet. Dev. Biol. 19, 1-11.
Lemonnier, J., Delannoy, P., Hott, M., Lomri, A., Modrowski, D. and Marie, P. J. (2000). The Ser252Trp fibroblast growth factor receptor-2 (FGFR-2) mutation induces PKC-independent downregulation of FGFR-2 associated with premature calvaria osteoblast differentiation. Exp. Cell. Res.
256, 158-167.
Lemonnier, J., Hay, E., Delannoy, P., Fromigue, O., Lomri, A., Modrowski, D. and Marie, P. J. (2001). Increased osteoblast apoptosis in Apert craniosynostosis: role of protein kinase C and interleukin-1. Am. J. Pathol.
158, 1833-1842.
Le Douarin, N. M., Ziller, C. and Couly, G. F.(1993). Patterning of neural crest derivatives in the avian embryo: in vivo and in vitro studies. Dev. Biol.
159, 24-49.
Liu, Y. H., Tang, Z., Kundu, R. K., Wu, L., Luo, W., Zhu, D., Sangiorgi, F., Snead, M. L. and Maxson, R. E.(1999). Msx2 gene dosage influences the number of proliferative osteogenic cells in growth centers of the developing murine skull: a possible mechanism for MSX2-mediated craniosynostosis in humans. Dev. Biol. 205, 260-274.
Lomri, A., Lemonnier, J., Hott, M., de Parseval, N., Lajeunie, E., Munnich, A., Renier, D. and Marie, P. J.(1998). Increased calvaria cell differentiation and bone matrix formation induced by fibroblast growth factor receptor 2 mutations in Apert syndrome. J. Clin. Invest. 101, 1310-1317.
Mansukhani, A., Bellosta, P., Sahni, M. and Basilico, C.(2000). Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J. Cell Biol. 149, 1297-1308.
Marie, P. J., Debiais, F. and Hay, E.(2002). Regulation of human cranial osteoblast phenotype by FGF-2, FGFR-2 and BMP-2 signaling. Histol. Histopathol. 17, 877-885.
McLeod, M. J.(1980). Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology22, 299-301.
Miki, T., Bottaro, D. P., Fleming, T. P., Smith, C. L., Burgess, W. H., Chan, A. M. and Aaronson, S. A. (1992). Determination of ligand-binding specificity by alternative splicing: two distinct growth factor receptors encoded by a single gene. Proc. Natl. Acad. Sci. USA89, 246-250.
Noden, D. M.(1983). The role of the neural crest in patterning of avian cranial skeletal, connective, and muscle tissues. Dev. Biol. 96, 144-165.
Oldridge, M., Zackai, E. H., McDonald-McGinn, D. M., Iseki, S., Morriss-Kay, G. M., Twigg, S. R., Johnson, D., Wall, S. A., Jiang, W., Theda, C. et al.(1999). De novo Alu-element insertions in FGFR2 identify a distinct pathological basis for Apert syndrome. Am. J. Hum. Genet. 64, 446-461.
Opperman, L. A.(2000). Cranial sutures as intramembranous bone growth sites. Dev. Dyn. 219, 472-485.
Ornitz, D. M. and Itoh, N.(2001). Fibroblast growth factors. Genome Biol.
2reviews3005.
Ornitz, D. M. and Marie, P. J. (2002). FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 16, 1446-1465.
Orr-Urtreger, A., Bedford, M. T., Burakova, T., Arman, E., Zimmer, Y., Yayon, A., Givol, D. and Lonai, P.(1993). Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2). Dev.
Biol. 158, 475-486.
Park, W. J., Theda, C., Maestri, N. E., Meyers, G. A., Fryburg, J. S., Dufresne, C., Cohen, M. M., Jr and Jabs, E. W.(1995). Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am. J. Hum. Genet. 57, 321-328.
Petiot, A., Ferretti, P., Copp, A. J. and Chan, C. T.(2002). Induction of chondrogenesis in neural crest cells by mutant fibroblast growth factor receptors. Dev. Dyn. 224, 210-221.
Pittenger, M. F., Mackay, A. M., Beck, S. C., Jaiswal, R. K., Douglas, R., Mosca, J. D., Moorman, M. A., Simonetti, D. W., Craig, S. and Marshak, D. R. (1999). Multilineage potential of adult human mesenchymal stem cells. Science 284, 143-147.
Rice, D. P., Aberg, T., Chan, Y., Tang, Z., Kettunen, P. J., Pakarinen, L., Maxson, R. E. and Thesleff, I.(2000). Integration of FGF and TWIST in calvarial bone and suture development. Development127, 1845-1855.
Richtsmeier, J. T., Baxter, L. L. and Reeves, R. H. (2000). Parallels of craniofacial maldevelopment in Down syndrome and Ts65Dn mice. Dev. Dyn. 217, 137-145.
Selleck, M. A., Scherson, T. Y. and Bronner-Fraser, M.(1993). Origins of neural crest cell diversity. Dev. Biol. 159, 1-11.
Slaney, S. F., Oldridge, M., Hurst, J. A., Moriss-Kay, G. M., Hall, C. M., Poole, M. D. and Wilkie, A. O.(1996). Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am. J. Hum. Genet. 58, 923-932.
von Gernet, S., Golla, A., Ehrenfels, Y., Schuffenhauer, S. and Fairley, J. D. (2000). Genotype-phenotype analysis in Apert syndrome suggests opposite effects of the two recurrent mutations on syndactyly and outcome of craniofacial surgery. Clin. Genet. 57, 137-139.
Wilkie, A. O., Slaney, S. F., Oldridge, M., Poole, M. D., Ashworth, G. J., Hockley, A. D., Hayward, R. D., David, D. J., Pulleyn, L. J., Rutland, P. et al.(1995). Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat. Genet. 9, 165-172.
Wilkinson, D. G. (1992). In Situ Hybridization: A Practical Approach.
London, UK: Oxford University Press.
Xu, X., Weinstein, M., Li, C., Naski, M., Cohen, R. I., Ornitz, D. M., Leder, P. and Deng, C. (1998). Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development125, 753-765.
Yu, K., Herr, A. B., Waksman, G. and Ornitz, D. M. (2000). Loss of fibroblast growth factor receptor 2 ligand-binding specificity in Apert syndrome. Proc. Natl. Acad. Sci. USA97, 14536-14541.
Yu, K., Xu, J., Liu, Z., Sosic, D., Shao, J., Olson, E. N., Towler, D. A. and Ornitz, D. M.(2003). Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130, 3063-3074.