Tuberculosis Outbreak in an Educational
Institution in Norway
Gunnstein Norheim,aSiri Seterelv,aTrude M. Arnesen,a
Anne Torunn Mengshoel,a,b Tone Tønjum,b,cJanne O. Rønning,a Vegard Eldholma
Norwegian Institute of Public Health, Oslo, Norwaya; Oslo University Hospital, Oslo, Norwayb; University of Oslo, Oslo, Norwayc
ABSTRACT Within 1 week in April 2013, three cases of pulmonary tuberculosis (TB) were reported among students attending training sessions at an educational institution in Oslo, Norway. By the end of October 2013, a total of nine epidemi-ologically linked cases had been reported. The outbreak encompassed a total of
24 cases from 2009 to 2014, among which all of the 22Mycobacterium
tuberculo-sis isolates available had identical mycobacterial interspersed repetitive-unit–
variable-number tandem-repeat (MIRU-VNTR) profiles (MtbC15-9 code 10287-189)
belonging to the Beijing lineage. Whole-genome sequencing (WGS) of the M.
tu-berculosisisolates revealed 20 variable nucleotide positions within the cluster, in-dicating a clonal outbreak. The most likely index case was identified and diag-nosed in Norway in 2009 but was born in Asia. WGS analyses verified that all of the cases were indeed part of a single transmission chain. However, even when combining WGS and intensified contact tracing, we were unable to fully recon-struct the TB transmission events.
KEYWORDS genotyping,Mycobacterium tuberculosis, Norway, outbreak, whole-genome sequencing
I
n 2013, 392 cases of tuberculosis (TB) were reported in Norway (7.8 cases per 100,000inhabitants) (1), defining it as a country with low incidence. A minority (13%) of these cases were reported among individuals born in Norway (1); their median age was 71 years. The TB incidence showed an all-time low in 1996, with 4.6 cases per 100 000 inhabitants. The annual incidence increased thereafter, mainly as a result of cases among individuals immigrating from countries with a high incidence of TB. There is, however, no indication that this significantly affects the incidence among Norwegian
citizens born in Norway. Molecular epidemiology studies based on IS6110restriction
fragment length polymorphism (RFLP) indicated that during 1994 to 2005, on average, 2 nonimmigrants and 13 immigrants per year developed the disease as a result of
infection within the country by importedMycobacterium tuberculosis(Mtb) (2). Similar
studies within the European Union/European Economic Area countries found that the TB incidence in a foreign-born population did not significantly affect the incidence of TB among the native population (2–4). However, these studies were based on RFLP, and the ability of this method to distinguish cases based on genetic homogeneity is limited. The National Reference Laboratory for Mycobacteria (NRL) implemented mycobacterial interspersed repetitive-unit–variable-number tandem-repeat (MIRU-VNTR) as a routine analysis for all isolates since 2011, and national guidelines require that all isolates be submitted to NRL. MIRU-VNTR is well standardized across the European Union, and many countries have implemented comprehensive schedules to utilize results to detect outbreaks and identify transmission chains. Whole-genome sequencing (WGS) is con-sidered to be a better tool for outbreak investigations due to its increased genomic resolution and may be able to utilize limited resources better and in a more targeted
Received6 June 2016Returned for
modification30 June 2016 Accepted3
February 2017
Accepted manuscript posted online15
February 2017
CitationNorheim G, Seterelv S, Arnesen TM,
Mengshoel AT, Tønjum T, Rønning JO, Eldholm V. 2017. Tuberculosis outbreak in an educational institution in Norway. J Clin Microbiol 55:1327–1333.https://doi.org/ 10.1128/JCM.01152-16.
EditorGeoffrey A. Land, Carter BloodCare &
Baylor University Medical Center
Copyright© 2017 American Society for
Microbiology.All Rights Reserved. Address correspondence to Gunnstein Norheim, gunnstein.norheim@fhi.no.
crossm
on May 16, 2020 by guest
http://jcm.asm.org/
manner than observed with MIRU-VNTR. Nonetheless, standardization of WGS and bioinformatics analyses for TB have a long way to go compared with MIRU-VNTR. In order to maximize the impact of WGS as part of routine surveillance, timeliness and close collaboration with outbreak teams is required, as are validated criteria for filtering single-nucleotide polymorphisms (SNPs) and interpretation of implications and meth-odological limits.
Here, we report the use of WGS to delineate an unusual outbreak detected in Oslo, Norway, in 2013 (5), primarily among Norwegian-born students attending physical exercise sessions at an educational institution. As part of the outbreak investigation, we linked epidemiological data from contact tracing to data from WGS of the causative Mtb isolates in an attempt to identify transmission patterns.
RESULTS
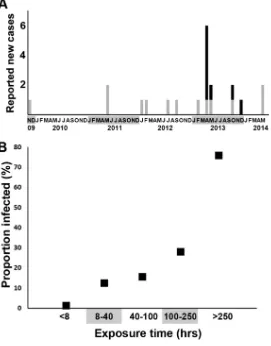
Outbreak at educational institution. The first patient recognized as part of the outbreak was a student at an educational institution in Oslo, Norway, who presented with TB symptoms for an extended period before diagnosis in April 2013 (5) (D1). Contact tracing around this case included 319 people, among which 8 were diagnosed with active TB. Forty-nine of the contacts were infected (defined as interferon gamma release assay [IGRA] positive), of whom 37 were subjected to preventive treatment. As expected, the proportion of infected individuals covaried with the degree of exposure (Fig. 1). Contact tracing performed for the 21 cases subsequent to the index case included 313 individuals; among these traced contacts, 2 were active TB cases but were among the same 21 cases. Of the 313 contacts, 30 were deemed infected, 12 of whom were subjected to preventive treatment. Cases were grouped as either linked to the educational institution (D cases) or not linked to the associated setting (S cases). Patients linked to the educational institution (median age, 22 years; range, 14 to 26 years) were significantly younger at the sampling date than those not linked to it
(median age, 36 years; range, 19 to 62 years) (P⫽0.0055).
Characteristics of outbreak isolates.All 22 cases presented with Mtb isolates that were high-level resistant to streptomycin (STR) but were susceptible to all other drugs tested. For 8 of the 9 patients linked to an educational institution, the country of birth of their parents was Norway or another country where TB has a low incidence, whereas for 8 of 13 (62%) of the nonlinked patients, the country of birth of their parents was one where TB is endemic. From 1 noncompliant case out of the 22, three isolates were cultivated over time (D1-1, D1-2, and D1-3), and the last isolate (D1-3) from 1 year after the first isolate was recognized had also become isoniazid (INH) resistant.
Outbreak defined by MIRU-VNTR.A total of 22 isolates with the same MIRU-VNTR profile (MtbC15-9 code 10287-189) were identified either as part of a cluster through routine analysis (20 cases) or by epidemiological linkage to a case diagnosed in 2011 (case S2) or 2009 (S1). Spoligotyping identified that the strains belonged to the Beijing-family (SIT type 1) (6), and 10 of these were identical in the 4-loci hypervariable MIRU-VNTR (7).
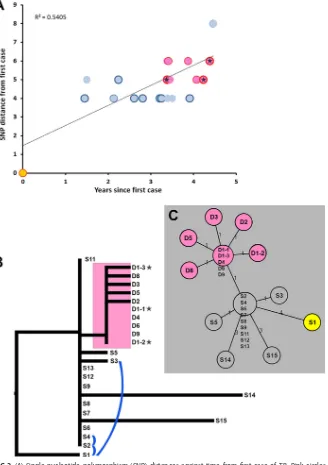
WGS.Pairwise comparison of the WGSs from the Mtb outbreak isolates revealed 20
SNPs separating the isolates sequenced over 5 years (Table 1, Fig. 2A). The median SNP difference between cases with a verified epidemiological link in this study was 1 (range, 0 to 5) (Fig. 2B). One isolate (S1) from an Asian-born patient diagnosed with TB in 2009 was found to be the most ancestral among the 22 isolates, indicating that this patient was the most likely index case of this outbreak. One particular SNP clearly separated the isolates associated with the educational institution (D) from all the other isolates (S) in the outbreak (Fig. 2C). The known epidemiological links between cases are shown in Fig. 2B, with the likely index case (D1) being the contact for 7 cases (D2 to D8). The mutation rate within the outbreak was estimated to be 1.1 SNP per genome per year (95% confidence interval [CI], 0.7 to 1.5).
Norheim et al. Journal of Clinical Microbiology
on May 16, 2020 by guest
http://jcm.asm.org/
DISCUSSION
The number of SNPs between subclusters observed in this study is within the range of those observed in other studies of cases with verified epidemiological links (Fig. 2C) (8, 9), and the mutation rate within the outbreak is somewhat higher than the mutation rate observed in a similar study in the United Kingdom (0.5 SNPs per genome per year; 95% CI, 0.3 to 0.7 SNPs per genome per year) (8). However, it is relatively high considering that Beijing family strains are known to be highly conserved (10).
The number of SNPs identified might vary according to the SNP filtering parameters applied (e.g., exclusion of repetitive genomic areas, coverage, minimum allele fre-quency for detection). The calling of multiple SNPs within short stretches of DNA might indicate mapping issues, but none of the SNPs identified in the current study were found within 10 bp of each other. Studies have proposed a range of 3 to 12 SNP difference as a significance threshold level for defining genomically linked confirmation in pairwise cases diagnosed with confirmed epidemiological contact (11, 12). The median SNP difference between cases with a verified epidemiological link in this study was 1 (range, 0 to 5). WGS thus clearly confirmed that the outbreak detected by means of MIRU-VNTR typing represented a single outbreak.
FIG 1(A) Epidemiological curve for the 22 cases with an available isolate, identified as part of the cluster defined by MIRU-VNTR typing, and their respective subcluster association identified based on whole-genome sequencing. D (black bars), patients associated with the educational-institution outbreak; S (gray bars), patients with no verified epidemiological association with the educational-institution outbreak setting. (B) Proportion infected among identified contacts of case D1. In total, 8 active cases and 49 cases of latent TB were newly diagnosed among contacts of case D1. (Adapted from reference 5 with permission of the publisher.) Exposure data:⬎250 h, 14 infected plus 5 active TB out of 25 contacts (76%); 100 to 250 h, 6 infected plus 1 active TB out of 25 contacts (28%); 40 to 99 h, 24 infected plus 1 active TB out of 153 contacts (16%); 8 to 39 h, 4 infected plus 1 active TB out of 40 contacts;⬍8 h, 1 infected and no active TB out of 76 contacts.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:3.585.69.340.74.420.2]The relatively high number of SNPs in cases S1, S14, and S15 was not fully explained; however, during follow-up, the medical staff experienced challenges with case S1 in regard to adherence to the treatment regimen. We have no information regarding similar adherence issues for cases S15 and S16. The number of SNP differences between these cases may also be explained by host-specific selection pressure factors (e.g., drug bioavailability and metabolism) or by the fact that some intermediate transmission hosts were not detected as part of this contact investigation.
Data from contact tracing indicated that case S1 was diagnosed within 1 month of arrival in Norway. Case S2 reported S1 as the likely source of infection, but S2 was not identified as part of the contact tracing for patient S1. The nine patients (S2, S4, S6, S7, S8, S9, S11, S12, and S13) diagnosed between May 2011 and October 2013 originated from a number of different countries. As these isolates were identical at the genome level, they were likely part of a single transmission chain, and that transmission occurred in Norway. Heterozygous alleles may be informative for linking cases in a transmission chain. We therefore studied in detail heterozygous SNP calls that did not meet the quality thresholds for inclusion in our genomewide SNP data set. All heterozy-gous calls were characterized by low sequencing depth in one or many isolates or by the presence of heterozygous calls in all or almost all of the isolates. These findings suggest that mapping errors are the likely cause of the heterozygous calls, and we thus chose not to study their distribution among patients in more detail.
The TB cases that were not associated with the educational institution were diag-nosed over 5 years, and few epidemiological links were identified among these patients (Fig. 2C). Since the cases were found to belong to a single outbreak, and the setting of transmission outside of the educational institution has not been identified, it is possible that not all of the cases in the outbreak since October 2009 have been identified. Two additional cases belonging to this outbreak were identified in 2015 (data not shown), and additional sporadic cases may surface in the years to come. It does seem likely, however, that the chain of transmission has been broken for the time being.
Three isolates from patient D1 (D1-1, D1-2, and D1-3), the suggested index case of the D cluster, that were isolated over 12 months were subjected to WGS. These isolates
were identical except for a single SNP in the promoter region ofinhA(⫺15 C¡T) in the
[image:4.585.40.551.82.317.2]last isolate (D1-3), conferring low-grade INH resistance (0.1 mg/liter) (Table 1). The
TABLE 1Single-nucleotide polymorphisms and indels identified in the study by WGS
Category Ref. positiona Gene Base substitution Classification Change
SNP used for phylogeny (n⫽20) 487700 pks6 C¡G Nonsynonymous A657G 771556 fadE8 C¡T Nonsynonymous L25F 885260 Rv0791c T¡C Nonsynonymous D194G 913711 phoY2 A¡G Synonymous
1120081 Rv1003 A¡G Synonymous 1224786 Rv1096 C¡T Synonymous
1473396 A¡T Intergenic
1495879 glgP G¡C Nonsynonymous G439A 1673425 C¡T Intergenic inhA⫺15 C¡T 1913093 mpg G¡A Nonsynonymous G39R
2082479 Rv1835c G¡A Nonsynonymous R37C 2531406 Rv2258c G¡T Synonymous
2844386 fas G¡A Synonymous
2899947 Rv2575 C¡T Synonymous
2903460 Rv2578c C¡A Nonsynonymous Q24H 3470232 ftsX A¡C Nonsynonymous L150V 3567950 Rv3197 A¡G Synonymous
3785688 Rv3371 G¡A Nonsynonymous G253S 4034342 Rv3592 C¡T Nonsynonymous L96F
4214469 G¡A Intergenic
Indel (n⫽2) 2522482 Rv2248 G¡del 1 Nonsynonymous M41fs 4099402 B11 ⫺¡ins G Noncoding RNA
aReference position relative to H37Rv genome.
Norheim et al. Journal of Clinical Microbiology
on May 16, 2020 by guest
http://jcm.asm.org/
combination of epidemiological and genomic data revealed pairwise distances of 0 to 1 SNP within isolates belonging to the educational-institution subcluster (D), whereas 1 to 5 SNPs were observed between this subcluster and other cases (Fig. 2C).
The TB outbreak described here is highly unusual in a Norwegian low-incidence setting in the last decades. The outbreak was caused by a Beijing lineage 2 strain that seems to be expanding globally (10). Despite a thorough clinical investigation, no epidemiological links were found between the educational-institution subcluster (D) and the rest of the outbreak (S). Links were also scarce within the S cluster, with the
FIG 2(A) Single-nucleotide polymorphism (SNP) distances against time from first case of TB. Pink circles indicate cases associated with the educational institution. Stars denote repeated isolates from the same patient (D1-1, D1-2, D1-3). (B) Maximum-likelihood phylogeny ofMycobacterium tuberculosis(Mtb) outbreak isolates. Blue links indicate known epidemiological links outside the D cluster. (C) Minimum-spanning network of Mtb outbreak isolates. The Mtb isolates belonging to the educational-institution students and their known epidemiological links are highlighted in pink. Stars are used to indicate the serial isolates from the single patient that is the likely index case of the suboutbreak at the educational institution. In A and C, the index case of the whole outbreak is highlighted in yellow.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:5.585.47.375.73.538.2]exception of the earliest cases, which were restricted to patients born in southern Asia. However, WGS confirmed that all of the MIRU-VNTR-linked cases belonged to a single clonal TB outbreak.
An effort to validate the completion of contact tracing, particularly among individ-uals born in countries with a high incidence of TB, may limit future transmission of TB within Norway. WGS can independently verify linked cases and transmission chain members; however, it can infer individual linkages only to a limited extent. The output of comparative genomics for Mtb complex strain resolution may be limited due to the relatively high sequence conservation and consequent redundancy in phylogeny. Further studies should address whether the availability of WGS-based linkage of Mtb cases can adequately guide the contact tracing to locations or settings where hidden cases are most likely to occur and thus lead to reduced transmission of TB.
MATERIALS AND METHODS
Culture, identification, and drug susceptibility testing ofM. tuberculosis.Samples were mainly cultured in the Bactec MGIT 960 system (Becton, Dickinson and Company [BD], Franklin Lakes, NJ) at the Department of Microbiology at Oslo University Hospital (OUS), except for samples from two patients. Apart from one sample derived from a knee biopsy specimen (S15), all samples were derived from sputum, bronchoalveolar lavage, or pleura fluid/biopsy samples. Isolates were primarily identified by using the MGIT TBc identification test (BD, USA), the Accuprobe TB assay (Gen-Probe, USA), or the Cobas TaqMan 48 PCR analyzer (Roche, USA). Mtb complex species identification was verified by GenoType MTBC line probe assay (Hain LifeScience, Nehren, Germany) at NRL, Norwegian Institute of Public Health (NIPH), which receives all new Mtb complex strains in Norway for identification, genotyping, and susceptibility testing when needed. Strains were tested for drug susceptibility against STR (4 mg/liter), INH (0.1 mg/liter), rifampin (1 mg/liter), ethambutol (5 mg/liter), and pyrazinamide (100 mg/liter) using the Bactec MGIT 960 system.
Genotyping.Routine 24-locus MIRU-VNTR testing (implemented since 2011) was performed using capillary electrophoresis and commercial kits (GenoScreen, Lille, France) (13) at NRL. The NRL, NIPH, has demonstrated 100% concordance in external proficiency testing organized by the National Institute for Public Health and the Environment (RIVM), The Netherlands, since its start. Matching MIRU-VNTR profiles were identified by a search of the MLVA Compare database (Ridom, Münster, Germany) containing all available national MIRU-VNTR data from Norway. Spoligotyping, which is based on the polymorphic direct-repeat region of the Mtb chromosome was performed on all 22 isolates (6), whereas a subset of 10 isolates were tested in the 4-loci hypervariable MIRU-VNTR (7).
WGS of M. tuberculosis DNA.WGS of the 22 isolates with the same MIRU-VNTR profile was performed at NRL. Briefly, DNA from the strains was extracted using E.Z.N.A. bacterial DNA isolation kits (Omega Bio-tek, Norcross, GA), following three cycles on a Precellys homogenizer (Bertin Corp., MD) at 6,300 rpm for 30 s. DNA sequencing libraries were prepared as previously described (14), and sequencing was performed on the Illumina MiSeq and NextSeq platform (Illumina, San Diego, CA), with all reads performed at 2⫻150 bp at a median depth of 352 (range, 52 to 706). The median pair distance was 221 bp (193 to 245 bp), and median template coverage was 99.14% to 99.21%.
Bioinformatics analyses.The Lasergene genomics suite (DNAStar, Madison, WI) was used to align sequencing reads to the H37Rv genome and subsequent identification of SNPs. The accession number of the H37Rv sequence used to align the reads wasNC_000962.3. SNPs located in or within 50 bp of regions annotated as PE/PPE genes, mobile elements, and repeat regions within 10 bp from each other, with a read depth of⬍8 or present in⬍75% of the reads in at least one isolate, were excluded from the analyses. We generally require a minimum sequencing depth of 8⫻for inclusion of SNPs in phylogenetic analyses, but due to high sequencing coverage, all of the SNPs that fulfilled the remaining quality score cutoffs were covered by a depth of⬎30⫻. Variable positions were concatenated to a single sequence per isolate and used for phylogenetic analyses. The proportion of the genome included after exclusion of problematic regions⫾50 bp was 90.1%. A maximum-likelihood (ML) phylogeny was computed in Seaview using a general-time-reversible model with four rate classes and a minimum-spanning network constructed using SeqSphere (Ridom) (Fig. 2B) (15). The ML tree was visualized and edited using Figtree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree). Path-O-Gen software (http://tree.bio.ed.ac.uk/software/ pathogen), which regresses the root-to-tip distance against the sampling date, was used to estimate the mutation rate within the TB outbreak. The ML phylogeny was rooted by including an unrelated Mtb Beijing isolate in the phylogenetic analyses (data not shown).
Ethical approval.Ethical approval was not required as the study was initiated within the legal mandate of NIPH to investigate and report on infectious disease outbreaks.
Accession number(s).Raw sequencing reads were deposited in the European Nucleotide Archive under study accession numberPRJEB12184.
ACKNOWLEDGMENTS
The research was funded by NIPH and OUS.
We thank Finn Bjørnar Jacobsen, Irena Szpinda, and Marie Noer at the OUS and Kari
Norheim et al. Journal of Clinical Microbiology
on May 16, 2020 by guest
http://jcm.asm.org/
Nilsen, Bente Forsdahl, Annika Reichmann, and Wibeke Kinander at NRL, NIPH, for excellent technical assistance.
We also thank primary laboratories in Norway submitting Mtb isolates to the NRL, NIPH.
REFERENCES
1. Arnesen TAHE, Mengshoel AT, Norheim G, Sandbu S, Winje BA. 2015. Tuberkulose i Norge 2014 —med behandlingsresultater for 2013. Nor-wegian Institute of Public Health, Oslo, Norway. https://www.fhi.no/ publ/2015/tuberkulose-i-norge-2014 —med-beha/.
2. Dahle UR, Eldholm V, Winje BA, Mannsaker T, Heldal E. 2007. Impact of immigration on the molecular epidemiology ofMycobacterium tubercu-losisin a low-incidence country. Am J Respir Crit Care Med 176:930 –935.
https://doi.org/10.1164/rccm.200702-187OC.
3. Sandgren A, Schepisi MS, Sotgiu G, Huitric E, Migliori GB, Manissero D, van der Werf MJ, Girardi E. 2014. Tuberculosis transmission between foreign- and native-born populations in the EU/EEA: a systematic review. Eur Respir J 43:1159 –1171.https://doi.org/10.1183/09031936.00117213. 4. Kamper-Jorgensen Z, Andersen AB, Kok-Jensen A, Kamper-Jorgensen M, Bygbjerg IC, Andersen PH, Thomsen VO, Lillebaek T. 2012. Migrant tuberculosis: the extent of transmission in a low burden country. BMC Infect Dis 12:60.https://doi.org/10.1186/1471-2334-12-60.
5. Arnesen TM, Seterelv S, Norheim G, Helgebostad SR, Mannsåker T, Ly IN, Rønning EJ, Steen TW. 2015. Tuberculosis outbreak in eastern Norway. Tidsskr Nor Laegeforen 135:2160 –2164. (In English, Norwegian)https:// doi.org/10.4045/tidsskr.14.0770.
6. Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Bunschoten A, Molhuizen H, Shaw R, Goyal M, van Embden J. 1997. Simultaneous detection and strain differentiation of Mycobacte-rium tuberculosisfor diagnosis and epidemiology. J Clin Microbiol 35: 907–914.
7. Allix-Beguec C, Wahl C, Hanekom M, Nikolayevskyy V, Drobniewski F, Maeda S, Campos-Herrero I, Mokrousov I, Niemann S, Kontsevaya I, Rastogi N, Samper S, Sng LH, Warren RM, Supply P. 2014. Proposal of a consensus set of hypervariable mycobacterial interspersed repetitive-unit-variable-number tandem-repeat loci for subtyping of Mycobacte-rium tuberculosisBeijing isolates. J Clin Microbiol 52:164 –172.https:// doi.org/10.1128/JCM.02519-13.
8. Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, Eyre DW, Wilson DJ, Hawkey PM, Crook DW, Parkhill J, Harris D, Walker AS, Bowden R, Monk P, Smith EG, Peto TE. 2013. Whole-genome sequencing to delineateMycobacterium tuberculosisoutbreaks: a retrospective ob-servational study. Lancet Infect Dis 13:137–146.https://doi.org/10.1016/ S1473-3099(12)70277-3.
9. Bryant JM, Schurch AC, van Deutekom H, Harris SR, de Beer JL, de Jager V, Kremer K, van Hijum SA, Siezen RJ, Borgdorff M, Bentley SD, Parkhill J, van Soolingen D. 2013. Inferring patient to patient transmission of
Mycobacterium tuberculosisfrom whole genome sequencing data. BMC Infect Dis 13:110.https://doi.org/10.1186/1471-2334-13-110.
10. Merker M, Blin C, Mona S, Duforet-Frebourg N, Lecher S, Willery E, Blum MG, Rusch-Gerdes S, Mokrousov I, Aleksic E, Allix-Beguec C, Antierens A, Augustynowicz-Kopec E, Ballif M, Barletta F, Beck HP, Barry CE, III, Bonnet M, Borroni E, Campos-Herrero I, Cirillo D, Cox H, Crowe S, Crudu V, Diel R, Drobniewski F, Fauville-Dufaux M, Gagneux S, Ghebremichael S, Hanekom M, Hoffner S, Jiao WW, Kalon S, Kohl TA, Kontsevaya I, Lille-baek T, Maeda S, Nikolayevskyy V, Rasmussen M, Rastogi N, Samper S, Sanchez-Padilla E, Savic B, Shamputa IC, Shen A, Sng LH, Stakenas P, Toit K, Varaine F, Vukovic D, Wahl C, Warren R, Supply P, Niemann S, Wirth T. 2015. Evolutionary history and global spread of the Mycobacterium tuberculosisBeijing lineage. Nat Genet 47:242–249. https://doi.org/10 .1038/ng.3195.
11. Roetzer A, Diel R, Kohl TA, Ruckert C, Nubel U, Blom J, Wirth T, Jaenicke S, Schuback S, Rusch-Gerdes S, Supply P, Kalinowski J, Niemann S. 2013. Whole genome sequencing versus traditional genotyping for investiga-tion of aMycobacterium tuberculosisoutbreak: a longitudinal molecular epidemiological study. PLoS Med 10:e1001387.https://doi.org/10.1371/ journal.pmed.1001387.
12. Walker TM, Lalor MK, Broda A, Saldana Ortega L, Morgan M, Parker L, Churchill S, Bennett K, Golubchik T, Giess AP, Del Ojo Elias C, Jeffery KJ, Bowler IC, Laurenson IF, Barrett A, Drobniewski F, McCarthy ND, Ander-son LF, Abubakar I, Thomas HL, Monk P, Smith EG, Walker AS, Crook DW, Peto TE, Conlon CP. 2014. Assessment ofMycobacterium tuberculosis
transmission in Oxfordshire, UK, 2007-12, with whole pathogen genome sequences: an observational study. Lancet Respir Med 2:285–292.
https://doi.org/10.1016/S2213-2600(14)70027-X.
13. Supply P, Allix C, Lesjean S, Cardoso-Oelemann M, Rusch-Gerdes S, Willery E, Savine E, de Haas P, van Deutekom H, Roring S, Bifani P, Kurepina N, Kreiswirth B, Sola C, Rastogi N, Vatin V, Gutierrez MC, Fauville M, Niemann S, Skuce R, Kremer K, Locht C, van Soolingen D. 2006. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing ofMycobacterium tuberculosis. J Clin Microbiol 44:4498 – 4510.https://doi.org/10.1128/JCM .01392-06.
14. Eldholm V, Norheim G, von der Lippe B, Kinander W, Dahle UR, Caugant DA, Mannsåker T, Mengshoel AT, Dyrhol-Riise AM, Balloux F. 2014. Evolution of extensively drug-resistantMycobacterium tuberculosisfrom a susceptible ancestor in a single patient. Genome Biol 11:490.https:// doi.org/10.1186/s13059-014-0490-3.
15. Gouy M, Guindon S, Gascuel O. 2010. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27:221–224. https://doi.org/10.1093/molbev/ msp259.