Aberrant promoter hypermethylation or histone modifications are recognized as an important mechanism in cancer initiation and progression.1These epigenetic
mechanisms are recognized as a “third pathway” in Knudson’s model of tumor-suppressor gene inactivation and can affect gene expression without accom-panying genetic changes.2DNA methylation occurs in the cytosine-guanine
sequence (CpG) in mammalian DNA strands. The pattern of methylation at cytosine residues in the CpG sequences is established during early development and is heritable.3In humans, approximately 70% of CpG dinucleotides are
methylated. The frequency of the CpG dinucleotide is relatively low in most of the human genome because of CG suppression. However, genomic sequencing reveals that CpG sites are unevenly distributed in the mammalian genome. About 60% of genes have 5’ promoter regions located in the CpG rich 0.3-2 kb stretches of DNA called CpG islands. CpG islands are normally unmethylated.4,5 Promoter
associated CpG islands need to be protected against DNA hypermethylation that results in irreversible inhibition of gene expression. In normal cells, this is evidenced by methylation associated silencing of imprinted genes and inactive X chromo-some genes. In cancers, this mechanism is utilized to inactivate tumor-suppressor genes.6
Histone modification is another key mechanism in transcriptional regulation, which is well conserved through a variety of species.7-9The less structured
N-terminal domains of all core histones protrude from the core histone and are subject to chemical modifications, such as acetylation, methylation, and phosphorylation at certain residues.10,11 The modification patterns of histone have been linked to
Epigenetic Cross-Talk between DNA Methylation and
Histone Modifications in Human Cancers
Yutaka Kondo
Division of Molecular Oncology, Aichi Cancer Center Research Institute, Nagoya, Japan.
DNA methylation, histone modifications, and the chromatin structure are profoundly altered in human cancers. The silencing of cancer-related genes by these epigenetic regulators is recognized as a key mechanism in tumor formation. Recent findings revealed that DNA methylation and histone modifications appear to be linked to each other. However, it is not clearly understood how the formation of histone modifications may affect DNA methylation and which genes are relevantly involved with tumor formation. The presence of histone modifications does not always link to DNA methylation in human cancers, which suggests that another factor is required to connect these two epigenetic mechanisms. In this review, examples of studies that demonstrated the relationship between histone modifications and DNA methylation in human cancers are presented and the potential implications of these epigenetic mechanisms in human neoplasia are discussed.
Key Words :Histone methylation, histone methyltransferase, DNA methylation
Received: June 3, 2009
Corresponding author: Dr. Yutaka Kondo, Division of Molecular Oncology, Aichi Cancer Center Research Institute, 1-1 Kanokoden, Chikusa-ku, Nagoya 464-8681, Japan. Tel: 81-52-764-2993, Fax: 81-52-764-2993312 3 E-mail: [email protected]
∙The author has no financial conflicts of interest.
© Copyright:
Yonsei University College of Medicine 2009
biological function and act as a “histone code”, which implies that transcription states can be predicted by simply
deciphering this code.8Recent studies showed that histone
modifications co-operate with DNA methylation to affect
gene inactivation in some tumor suppressor genes.12-16DNA
methylation and histone modifications might interact with each other and establish stable gene silencing. In addition to this interaction, histone H3 lysine 27 trimethylation (H3K27me3), which is involved in the regulation of home-otic (Hox) gene expression and in the early steps of X-chromosome inactivation in women, can be solid silencing machinery that is independent of DNA methylation in
human cancers.17-19
Although evidence for aberrant DNA methylation and histone modification changes in cancers is accumulating, how aberrant DNA methylation and histone modifications interact in a certain loci, why some genes are particular targets of epigenetic machinery, and whether aberrant DNA methylation and histone modifications confer specific roles in cancer initiation and progression remains unclear.
In this review, we discuss the current understanding of gene silencing by DNA methylation and histone modifica-tions, focusing especially on histone H3 lysine 9 methyla-tion (H3K9me) and H3K27me3, as related to human cancers.
Histone modifications
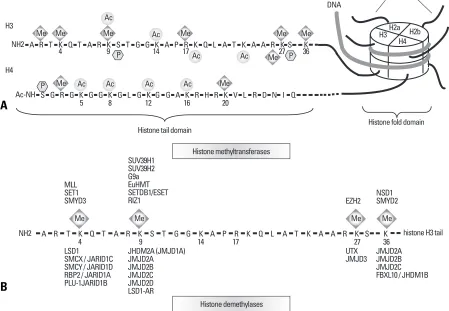
Nucleosomes consist of 147 bp DNA wrapped around the core histone that comprises histone proteins, H2A, H2B,
H3, and H4 (Fig. 1A).7,20The N-terminal domains of all
core histones are subject to chemical modifications, such as acetylation, methylation, ubiquitylation, and
phosphory-lation at certain residues.8Histone modifying enzymes
bring the complexity of post-translational modifications that can either activate or repress transcription, depending on the type of chemical modification and its location in the histone protein. The modification pattern of histone has been linked to chromatin structure and gene function
during development as well as tumorigenesis.6,10,11
These different combinations of histone tail
modifica-4 9 14 17 27 36
Me Me Me Me
NH2 A R T K Q T A R K S T G G K A P R K Q L A T K A A R K S K histone H3 tail
MLL SET1 SMYD3
SUV39H1 SUV39H2 G9a EuHMT SETDB1/ESET
RIZ1 EZH2
NSD1 SMYD2
LSD1
SMCX / JARID1C SMCY / JARID1D RBP2 / JARID1A PLU-1JARID1B
JHDM2A (JMJD1A) JMJD2A JMJD2B JMJD2C JMJD2D LSD1-AR
UTX JMJD3
JMJD2A JMJD2B JMJD2C FBXL10 / JHDM1B Histone methyltransferases
Histone demethylases
4 9 14 17 27 36
Me Me Me
Me Me
Me Me Ac
Ac
Ac Ac
P P
NH2 A R T K Q T A R K S T G G K A P R K Q L A T K A A R KS K
Ac-NH S G R G K G G K G L G K G G A K R H R K V L R D N I Q Ac
P Me Ac Ac Ac Me
5 8 12 16 20
Histone tail domain H3
H4
H3 H4
H2a H2b
Histone fold domain DNA
[image:2.595.60.509.340.651.2]Histone tail
Fig. 1.Schematic of histone modifications and modifiers. (A) DNA is compacted in the nucleus. The fundamental repeating unit of chromatin is the nucleosome, which consists of 147 base pairs of DNA wrapped around an octamer of the following core histone proteins: H2A, H2B, H3, and H4. Histone proteins consist of less structured amino-terminal tails (tail domain) that protrude from the nucleosome and globular carboxy-terminal domains make up the nucleosome scaffold (fold domain). The histone tail domain could be a target of a variety of post-translational modifications, including acetylation (Ac), methylation (Me) and ubiquitination of lysine (K) residues, phosphorylation (P) of serine (S) and threonine (T) residues, and methylation of arginine (R) residues. (B) Lysine residues in the histone H3 tail are the targets of methyltransferases and demethylases. Histone methylase, MLL, myeloid/lymphoid or mixed-lineage leukemia; SMYD3, SET and MYND domain containing 3; RIZ1, retinoblastoma protein-interacting zinc-finger protein 1; EZH2, enhancer of zeste homologue 2; NSD1, Nuclear receptor binding SET domain protein 1. Histone demethylases, LSD1, lysine-specific histone demethylase 1; SMCX, Smcx homolog, X chromosome; SMCY, Smcy homolog, Y chromosome.42
A
tions influence transcription by affecting chromatin
struc-tures.8Modifications on the lateral surface of the core
histone could also affect histone-DNA interactions as well as N-terminal domains. The respective enzymes vary in their potential to induce mono-, di-, or tri-methylation (Fig. 1B). Since lysine methylation is more stable than other modifications, there has been until recently, discussion as to why stably methylated histone lysine residues contri-buted to the establishment and propagation of different
patterns of gene expression in the same genome.21Recent
comprehensive reports revealed that histone methylation is reversible by demethylases (Fig. 1B). Given that histone methylation has important roles in development and other biological events, including tumorigenesis, histone deme-thylases might be key molecules in cellular processes.
Generally, acetylation of lysine residues on histone H3 and H4 leads to the formation of an open chromatin
struc-ture, which indicates transcriptional activity.22In contrast,
methylation on lysine residues link to either activation or
repression depending on its location.11 Trimethylation at
K9 on histone H3 (H3K9me3) or K20 on histone H4 (H4K20me3) has been shown to form heterochromatin from yeast to humans. Dimethylation at K9 (H3K9me2) is associated with inactivation of gene expression at the euchromatic region. Trimethylation at histone H3K27 (H3K27me3) is a distinct histone modification involved in the regulation of homeotic (Hox) gene expression and in
the early steps of X-chromosome inactivation in women.23
This process is mechanistically linked to polycomb group (PcG) proteins. PcG proteins, an enhancer of zeste 2 (EZH2), which is a member of polycomb repressor complex 2 (PRC2), has histone methyltransferase activity with substrate specificity for H3K27. H3K27me3 serves as a signal for specific binding of the chromodomain of another polycomb repressor complex, PRC1, which includes B lymphoma Mo-MLV insertion region 1 (BMI-1), ring
K4 Me
K27 Me
K27 Me
K4 Me
K27 Me K4
Me
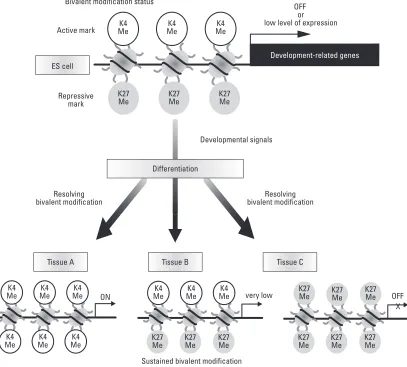
OFF or low level of expression Bivalent modification status
Active mark
Repressive mark ES cell
Differentiation
Developmental signals
K4 Me
K4 Me K4
Me
Sustained bivalent modification very low Tissue A
Development-related genes
K4 Me
K4 Me K4
Me
Tissue B Tissue C
K4 Me
K4 Me K4
Me
K27 Me
K27 Me
K27 Me K27
Me K27Me
K27 Me
K27 Me
K27 Me
K27 Me
OFF X ON
Resolving bivalent modification Resolving
[image:3.595.105.512.318.685.2]bivalent modification
finger protein 1 (RING1), human polycomb (HPC), and
human polyhomeotic homologue (HPH).24The binding of
PRC1 blocks the recruitment of transcriptional activation factors, such as SWI/SNF, and the presence of PRC1 prevents initiation of transcription by RNA polymerase II.25,26 Di- or tri-methylation on K4 on histone H3
(H3K4me2/me3) localizes to sites of active transcription and this modification may be stimulatory for transcription. Several recent studies indicated the existence of ‘bivalent’ domains consisting of dual marks of repressive histone H3K27me3 and the activation of histone H3K4me3 modifications on development-associated genes that are silent in ES cells and activated during differentiation (Fig. 2).27,28The dynamic alteration of lysine methylation
contributes to reversible and plastic regulation of gene expression in varieties of the cellular process, which contrasts with stable gene inactivation by DNA methylation.
DNA methylation in cancer
Promoter DNA methylation is the most widely studied
epigenetic modification of human cancers.29Compared to
normal tissues, cancer DNA shows global hypomethylation and hypermethylation at gene-associated CpG islands that are normally unmethylated. Cancer hypomethylation is found in repetitive elements localized in satellite sequences or pericentromeric regions and has been reported to lead to
genomic instability.30Hypermethylation in CpG islands is
found in several types of cancers and recognized as a mechanism of tumor suppressor gene silencing. Initial studies indicated DNA methylation as a cause of tumori-genesis. For example, inactivation of the cyclin-dependent kinase inhibitor P16/CDKN2A by methylation leads to the disruption of cell-cycle regulation and potentially provides
a growth advantage to affected cells.31Another tumor
suppressor genes, P14/ARF is also a target of inactivation
by DNA methylation.32 P14/ARF activates P53 through
interactions with MDM2, and losses of P14/ARF negati-vely affect P53 function. Inactivation by methylation has also been found in DNA repair genes, such as hMLH1 and
MGMT.33,34Tumor suppressor gene silencing by DNA
methylation promotes cell proliferation and may provide strong selective advantages. However, potential onco-genes, such as COX2 and hTERT, are also targets of aberrant DNA methylation, which illustrates the point that
methylation is initially independent of gene function.35,36
These data suggest that cancer-associated DNA methy-lation is not restricted to tumor suppressor genes and that the profile of methylation in cancers may not simply be a result of selection.
Recent accumulating evidence suggests that DNA methy-lation is a late step in the gene silencing process, even
though such methylation has been detected in pre-cancerous tissues. The establishment of fundamental DNA methy-lation patterns during early development might be
accomp-lished through histone modification.37H3K4me has recently
been suggested to protect gene promoters from de novo
DNA methylation in somatic cells.38,39Therefore, de novo
methylation is closely associated with the removal of methylation from H3K4 by Lsd1 and jmj-C containing demethylases [Jarid 1a and Jarid 1b, also known as K demethylases 5A (KDM5A) and K (lysine)-demethylases 5B (KDM5B)] on certain loci.40-42After
removal of methylation from H3K4, Dnmt3A/Dnmt3B-Dnmt3L complexes can target these loci, resulting in de novo methylation.37,43However, H3K4me marks most CpG
islands, since RNA polymerase II recruits specific H3K4 methyltransferases, which link to CpG islands of actively transcribed genes.39,44 Contact between DNMT3L and
nucleosomes are inhibited by H3K4me. As a result, most CpG is prevented from de novomethylation.
The DNA methylation pattern is maintained by DNA methyltransferase 1 (Dnmt1), which is associated with a replication complex. Studies have indicated that Dnmt1, together with UHRF1 (also known as Np95 or ICBP90), recognizes the hemimethylated CpG residues and methylates the opposite strand, resulting in a faithful copy of the methylation profile of the parent cell.45Regulation of
Dnmt1 methylation by histone demethylase Lsd1 and histone methylase Set7/9 has recently been reported.46
According to this model, Lsd1counteracts methylation of Dnmt1 by Set7/9, which results in the stabilization of Dnmt1 and enables DNA methylation to be maintained.
Once DNA methylation has been established on CpG islands, this modification is generally irreversible without artificially altering key factors in the cells, as was shown in iPS cells.47,48Therefore, facultative regulation by histone
modifications is stably determined by DNA methylation on the silenced loci in cancer cells.
Interrelation between DNA methylation and histone modifications
Early studies have shown that the link between DNA methylation and histone modifications is mediated by a group of proteins with methyl DNA binding activity, including methyl CpG binding protein 2 (MeCP2), Methyl-CpG binding domain protein 1 (MBD1), and Kaiso [also known as ZBTB 33 (Zinc finger and BTB domaincon-taining protein 33)]. These proteins localize to DNA methylated promoters and recruit a protein complex that contains histone deacetylases (HDACs) and histone methyltransferases.49-51These studies suggest DNA
methy-lation inhibits H3K4me, which is also evidence that DNA
methylation affects histone modifications.38,39 However,
early studies in fungi (Neurospora crassa) show that
mutations of histone H3K9 methyltransferase reduced DNA methylation, indicating a simple linear model in which H3K9 methylation acts as an upstream epigenetic
mark that signals to DNA methylation.52In embryonic stem
cells, Oct3/4 is inactivated after lineage commitment. For this silencing process, a repressor complex is recruited that contains histone methyltransferase G9a and enzymes with histone deacetylase activity. Subsequently,
methyltrans-ferase, DNMT3A, and DNMT3b, which catalyze de novo
DNA methylation, are recruited at the promoter.13
Intri-guingly, interaction between G9a and the DNMTs depends
on the ankyrin motif of G9a.53By contrast, the SET domain,
which is responsible for the methyltransferase activity of G9a, does not interact with DNMTs. Indeed, mutation of the SET domain is inert in methyltransferase activity and disrupts H3K9 methylation without affecting DNA
methy-lation.54,55These data suggest that DNA methylation on the
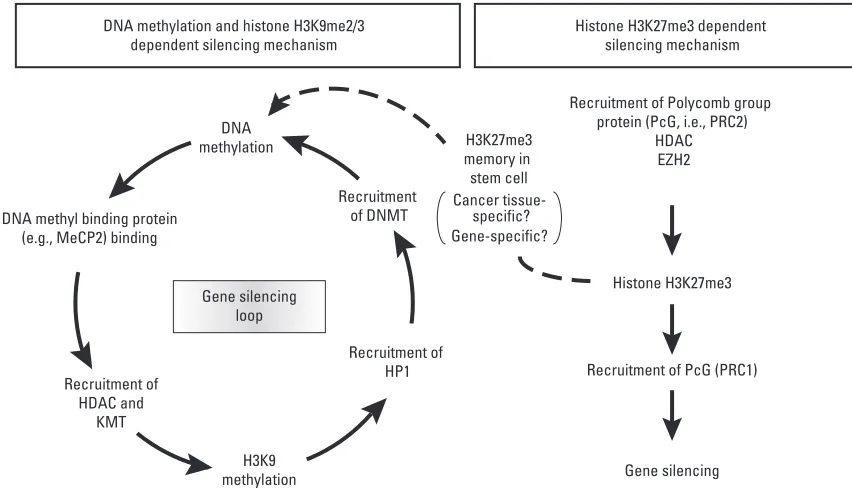
promoter depends on the recruitment of G9a (especially ankyrin motif), rather than the histone methyltransferase activity itself. The interactions between DNA methylation and histone H3K9 methylation currently fit a model whereby these two changes form a reinforcing silencing loop or bidirectional crosstalk, and this may explain why silencing is less stable in organisms that lack DNA
methy-lation (Fig. 3).2
Recently, links between PcG-mediated methylation on
H3K27 and de novoDNA methylation in cancers were
described using ChIP analyses coupled with bioinformatic database mining, which supports the conventional view that the mark imposed by PRC2 during development may
predispose some genes for later de novomethylation (Fig.
3).14-16Genome-wide analysis combined with methylation
prediction models revealed that CpG islands predicted to be methylation-prone revealed a strong association with embryonic targets of PRC2 and a subset of PRC2 targets
that were more likely to be hypermethylated in cancer.56
Biochemical study also showed that DNMTs bind to EZH2, however, this interaction seems to be detected only
under certain conditions.48,57,58Obviously, the presence of
histone methylation at H3K9 or H3K27 does not always
lead to de novomethylation. Indeed, subsets of PcG target
genes are methylated in one cancer, even though these same genes are completely unmethylated in another cancer from the same normal tissue origins. Additional factors are required for triggering DNA methylation in the genes enriched with those histone modifications.
More recently, comprehensive genome-wide and func-tional analysis revealed that PcG-mediated H3K27me3 is a mechanism of tumor-suppressor gene silencing in cancer that is potentially independent of promoter DNA methy-lation and a direct interaction between PcG-mediated
DNA methylation and histone H3K9me2/3 dependent silencing mechanism
Recruitment of Polycomb group protein (PcG, i.e., PRC2)
HDAC EZH2
Histone H3K27me3
Recruitment of PcG (PRC1)
Gene silencing Histone H3K27me3 dependent
silencing mechanism
DNA methylation
Recruitment of DNMT
Recruitment of HP1
H3K9 methylation Recruitment of
HDAC and KMT DNA methyl binding protein
(e.g., MeCP2) binding
H3K27me3 memory in stem cell
Gene silencing loop
[image:5.595.94.520.429.673.2]Cancer specific? Gene-specific?
methylation on H3K27 and de novoDNA methylation
pathways is unlikely in cancers.18,19This independence of
DNA-methylation and histone marking appear to conflict with previous studies. It must be noted that the majority of genes enriched with repressive mark H3K27me3 in prostate cancers do not have the CpG island in the promoter regions, whereas genes targeted by PcG complexes are generally
associated with CpG island promoters in ES cells.18,48 This
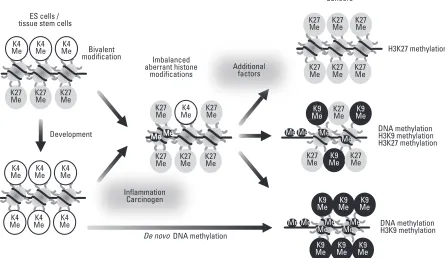
indicates that targets of PcG-mediated methylation on H3K27 among ES cells, normal tissue cells, and cancer cells are different and that cancer cells usurp the silencing mecha-nisms to extinguish functional pathways by H3K27me3. Now, three lines might be considered in the silencing machinery associated with PcG-mediated methylation on
H3K27 (Fig. 4). First, genes are de novo repressed by
PcG-mediated methylation on H3K27, which supports the observation of distinct target genes between normal and cancer cells (particularly targeting non-CpG island promo-ters). Second, during early tumorigenesis, subsets of genes become methylated, and a large portion of these are initially marked by the PcG complex (targeting CpG island promoters). In this case, several genes undergo epigenetic programming, in which genes that are originally silenced by PcG acquire DNA methylation as an alternative silencing
mechanism. This epigenetic switch to DNA methylation-mediated repression reduces the epigenetic plasticity, locking the silencing of key regulators and contributing to
tumorigenesis.19Third, genes that are originally silenced by
PcG acquire DNA methylation to some extent. In this case, H3K27me3 and DNA methylation co-exist on the same promoter and PcG-mediated H3K27me3 is the dominant silencing machinery, which can be reactivated by inhibition of PRC complexes. In cancers it appears that different forms of gene silencing contribute to tumorigenesis, which range from a flexible and plastic repressor-based mechanism to a highly stable inactivation that is maintained by DNA methylation.
Future direction: perspectives on epigenetic therapy
This review has focused on the relationship between two major epigenetic mechanisms: DNA methylation and histone methylation. Clarifying this relationship is impor-tant in the context of cancer epigenetic therapy. Restoring gene function silenced by epigenetic changes in cancer has the potential of ‘normalizing’ cancer cells by epigenetic therapy. This could have a serious impact on the prevention and treatment of human cancers. However, there are important questions that must be understood in regard to
K4 Me
K27
Me K27Me
K4 Me
K27 Me K4
Me
Inflammation Carcinogen
K27
Me K27Me K27Me
M K27 Me
Me Me Me Me
K27 Me K9
Me MeK9
K27
Me MeK9 K27Me Cancers
Additional factors
K9
Me MeK9 MeK9
K9
Me MeK9 MeK9
DNA methylation H3K9 methylation DNA methylation H3K9 methylation H3K27 methylation K27
Me K27Me
K27
Me K27Me K27Me
H3K27 methylation
K27 Me
K4
Me MeK4
K4 Me
K4
Me MeK4
K4 Me
Development
Imbalanced aberrant hisstone
modificatioons
tion DNA methylat De novo
ES cells / tissue stem cells
Bivalent modification
Me Me
Me Me
[image:6.595.73.518.387.645.2]MeMe MeMe
human cancers and how DNA methylation affects chro-matin structure and vice versa remain. It will be important to elucidate the precise role of these epigenetic mechanisms that allow cancer cells to revert to a more normal state through epigenetic reprogramming.
HDAC inhibitors lead to the accumulation of acetylation in histones, which results in reversion of chromatin status
and transcriptional activity to a normal state.59HDAC
inhi-bitors have been shown to induce P21, which is responsible for cell cycle arrest and cell differentiation. Another possible role of HDAC inhibitors in antitumor activity might be to inhibit PcG proteins, some of which have been recognized as important and uniquely acting oncogenic proteins, in cancers. Since PcG-mediated gene silencing is initiated by the HDAC activity of PRC2, inhibition of HDAC can efficiently reactivate the H3K27me3 target genes.18,60
DNA methylation is a therapeutic target of human
cancers.59,61 The cytosine analogues, azacytidine and
5-aza-2’-deoxycytidine, are powerful inhibitors of DNA methylation, which are incorporated into DNA during cell division. They trap DNA methyltransferases and lead to cell differentiation and growth repression. These demethy-lating agents have received FDA approval for the treatment of myelodysplastic syndrome. However, transiently induced demethylation is impaired after the withdrawal of chemicals,
which has been suggested in cell line studies.62This
pheno-menon might be partially explained by epigenetic memory retained in chromatin, which may include histone methyla-tion and interfere with the complete epigenetic reprogram-ming in cancer cells, as shown in this review. Combining DNA methyltransferase and histone methyltransferase inhibitors might provide a clue to solving this incomplete reprogramming. In addition, the synergistic effects between DNA methyltransferase and HDAC inhibitors suggest clinical trials of this approach to restore gene function
silenced by aberrant chromatin changes in cancers.63
Nevertheless, complete understanding of these epigenetic modifications and their cross-talk will lead to the develop-ment of the most effective therapies possible. With such knowledge of epigenetics at our disposal, we may yield the full capacity to not only treat but to prevent cancer. By correcting epigenetic abnormalities that predispose to cancers, the promise for epigenetic therapies as an essential treatment option will be achieved.
1. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002;3:415-28.
2. Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer
Metastasis Rev 2004;23:29-39.
3. Turker MS. The establishment and maintenance of DNA methy-lation patterns in mouse somatic cells. Semin Cancer Biol 1999; 9:329-37.
4. Bestor TH, Gundersen G, KolstøAB, Prydz H. CpG islands in mammalian gene promoters are inherently resistant to de novo methylation. Genet Anal Tech Appl 1992;9:48-53.
5. Cross SH, Bird AP. CpG islands and genes. Curr Opin Genet Dev 1995;5:309-14.
6. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer 2006;6:107-16.
7. Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999; 98:285-94.
8. Strahl BD, Allis CD. The language of covalent histone modifi-cations. Nature 2000;403:41-5.
9. Bird AP, Wolffe AP. Methylation-induced repression--belts, braces, and chromatin. Cell 1999;99:451-4.
10. Cheung P, Allis CD, Sassone-Corsi P. Signaling to chromatin through histone modifications. Cell 2000;103:263-71.
11. Jenuwein T, Allis CD. Translating the histone code. Science 2001;293:1074-80.
12. Kondo Y, Shen L, Yan PS, Huang TH, Issa JP. Chromatin immu-noprecipitation microarrays for identification of genes silenced by histone H3 lysine 9 methylation. Proc Natl Acad Sci U S A 2004;101:7398-403.
13. Feldman N, Gerson A, Fang J, Li E, Zhang Y, Shinkai Y, et al. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nat Cell Biol 2006;8:188-94. 14. Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L,
et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silenc-ing. Nat Genet 2007;39:237-42.
15. Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, et al. Epigenetic stem cell signature in cancer. Nat Genet 2007;39:157-8.
16. Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 2007;39:232-6.
17. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002;298:1039-43.
18. Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C, et al. Gene silencing in cancer by histone H3 lysine 27 trimethy-lation independent of promoter DNA methytrimethy-lation. Nat Genet 2008;40:741-50.
19. Gal-Yam EN, Egger G, Iniguez L, Holster H, Einarsson S, Zhang X, et al. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A 2008;105:12979-84.
20. Luger K, Richmond TJ. The histone tails of the nucleosome. Curr Opin Genet Dev 1998;8:140-6.
21. Trojer P, Reinberg D. Histone lysine demethylases and their impact on epigenetics. Cell 2006;125:213-7.
22. Roth SY, Allis CD. Histone acetylation and chromatin assembly: a single escort, multiple dances? Cell 1996;87:5-8.
14:155-64.
24. Levine SS, Weiss A, Erdjument-Bromage H, Shao Z, Tempst P, Kingston RE. The core of the polycomb repressive complex is compositionally and functionally conserved in flies and humans. Mol Cell Biol 2002;22:6070-8.
25. Shao Z, Raible F, Mollaaghababa R, Guyon JR, Wu CT, Bender W, et al. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell 1999;98:37-46.
26. Dellino GI, Schwartz YB, Farkas G, McCabe D, Elgin SC, Pirrotta V. Polycomb silencing blocks transcription initiation. Mol Cell 2004;13:887-93.
27. Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006;125:315-26.
28. Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Gian-noukos G, et al. Genome-wide maps of chromatin state in pluri-potent and lineage-committed cells. Nature 2007;448:553-60. 29. Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007;
128:683-92.
30. Cadieux B, Ching TT, VandenBerg SR, Costello JF. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate red-uctase allele status, and increased proliferation. Cancer Res 2006;66:8469-76.
31. Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, et al. Inactivation of the CDKN2/p16/MTS1 gene is freq-uently associated with aberrant DNA methylation in all common human cancers. Cancer Res 1995;55:4525-30.
32. Robertson KD, Jones PA. The human ARF cell cycle regulatory gene promoter is a CpG island which can be silenced by DNA methylation and down-regulated by wild-type p53. Mol Cell Biol 1998;18:6457-73.
33. Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 1997;57: 808-11.
34. Costello JF, Futscher BW, Tano K, Graunke DM, Pieper RO. Graded methylation in the promoter and body of the O6-methyl-guanine DNA methyltransferase (MGMT) gene correlates with MGMT expression in human glioma cells. J Biol Chem 1994; 269:17228-37.
35. Toyota M, Shen L, Ohe-Toyota M, Hamilton SR, Sinicrope FA, Issa JP. Aberrant methylation of the Cyclooxygenase 2 CpG island in colorectal tumors. Cancer Res 2000;60:4044-8. 36. Devereux TR, Horikawa I, Anna CH, Annab LA, Afshari CA,
Barrett JC. DNA methylation analysis of the promoter region of the human telomerase reverse transcriptase (hTERT) gene. Cancer Res 1999;59:6087-90.
37. Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methy-lation of DNA. Nature 2007;448:714-7.
38. Okitsu CY, Hsieh CL. DNA methylation dictates histone H3K4 methylation. Mol Cell Biol 2007;27:2746-57.
39. Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007;39:457-66.
40. Godmann M, Auger V, Ferraroni-Aguiar V, Di Sauro A, Sette C, Behr R, et al. Dynamic regulation of histone H3 methylation at
lysine 4 in mammalian spermatogenesis. Biol Reprod 2007; 77:754-64.
41. Hotz HR, Peters AH. Protein demethylation required for DNA methylation. Nat Genet 2009;41:10-1.
42. Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, et al. New nomenclature for chromatin-modifying enzymes. Cell 2007;131:633-6.
43. Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 2007;449:248-51.
44. Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007;130:77-88.
45. Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007;450:908-12. 46. Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet 2009;41:125-9.
47. Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hoche-dlinger K, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 2007;448:318-24.
48. Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature 2008;454:49-55.
49. Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998;393:386-9.
50. Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol 1998;18:6538-47.
51. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002;16:6-21.
52. Tamaru H, Selker EU. A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature 2001;414:277-83. 53. Epsztejn-Litman S, Feldman N, Abu-Remaileh M, Shufaro Y,
Gerson A, Ueda J, et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol 2008;15:1176-83.
54. Dong KB, Maksakova IA, Mohn F, Leung D, Appanah R, Lee S, et al. DNA methylation in ES cells requires the lysine methyltran-sferase G9a but not its catalytic activity. EMBO J 2008;27:2691-701.
55. Tachibana M, Matsumura Y, Fukuda M, Kimura H, Shinkai Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J 2008;27:2681-90. 56. McCabe MT, Lee EK, Vertino PM. A multifactorial signature of
DNA sequence and polycomb binding predicts aberrant CpG island methylation. Cancer Res 2009;69:282-91.
57. ViréE, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006;439:871-4.
58. Fouse SD, Shen Y, Pellegrini M, Cole S, Meissner A, Van Neste L, et al. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell 2008;2:160-9. 59. Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present
and future. Nat Rev Drug Discov 2006;5:37-50.
the human polycomb-group protein EED involves histone deace-tylation. Nat Genet 1999;23:474-8.
61. Issa JP. Decitabine. Curr Opin Oncol 2003;15:446-51.
62. McGarvey KM, Fahrner JA, Greene E, Martens J, Jenuwein T, Baylin SB. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin
state. Cancer Res 2006;66:3541-9.