Journal of Chemical and Pharmaceutical Research, 2017, 9(2):176-180
Research Article
CODEN(USA) : JCPRC5
ISSN : 0975-7384
176
Conformational Properties of N-Formyl-L-Cys-NH
2in Its
D-Backbone
Conformations as Computed by Semi-Empirical and
Ab Initio
Methods
Malika Bourjila
*, Rachida Tijar, Brahim El Merbouh, Anouar El Guerdaoui, Rachid
Drissi El Bouzaidi, Abderrahmane El Gridani and Mohamad El Mouhtadi
Department of Chemistry, Faculty of Science, B.P. 8106, Ibn Zohr University, 80000, Agadir, Morocco _______________________________________________________________________
ABSTRACT
A conformational study on the preferred minima D (C7ax)of N-For-L-Cys-NH2 in isolated state was carried out
using multi niche crowding genetic algorithm coupled with semi empirical-method AM1 (AM1/GA-MNC). All conformations located were subjected to geometry optimization at the HF/3-21G and the HF/6-31G++ (d, p) levels of theory. Six of the nine side-chain conformers were located and are ɣD[g-, g+], ɣD [g-, g-], ɣD[g-, a],
ɣD[a, g-], ɣD[a, g+] and ɣD[g+, g-], no conformational migrations were observed. All these structures are
stabilized by the hydrogen bond associated with the seven-member ring of the ɣ backbone conformation C=O…H-N. Two conformers had side chain-backbone intramolecular hydrogen bonding S-H…O=C at AM1/GA-MNC and HF/3-21G.
Keywords: Genetic algorithm; Semi-empirical; Ab initio; Cysteine; Conformational space
_____________________________________________________________________________
INTRODUCTION
Characteristics of Ramachandran map of trans N-For-L-Ser-amide have already been studied extensively [1-5], while the informations about conformational analysis of potential energy surface of L-Cysteine diamides are limited [6-8]. A preliminary work has already analyzed the potential energy surface (PES) of neutral and protonated glycine using the AM1/GA-MNC method [9]. In the present study, we wish to explore the whole conformational space of N-For-L-Cys-NH2 in isolated state and carried out all D conformations using our
computing calculation technique, AM1/AG-MNC. The equilibrium structures obtained had been re-optimized by ab initio calculation (HF/3-21G, HF/6-31G++(d, p)). In scheme 1, are represented the numbering system and the torsional angles definition adopted in this study.
Scheme1: The atomic numbering adopted in this study
Computational method
we carried out a conformational study of potential energy surface E ( χ1, χ2) of N-For-L-Cys-NH2 using
177
six side chain conformers located were subjected to geometry optimization at two levels of theory: HF/3-21G and the HF/6-31G++ (d, p), using the Gaussian 09 [10]. Torsional angles were specified within -180° and 180° for both backbone (,) and side-chain (χ1, χ2) conformations. All the relative energies are given in kcal/mol using the conversion factor 1 hartree = 627.5 kcal/mol. Three stable orientations can be found for χ1 and χ2; gauche + (g+), anti (a) and gauche –(g-).
RESULTS AND DISCUSSION
Limiting our considerations only to trans-peptide bonds, = O6-C5-N4-C3 ≈0 °. The potential energy surface is
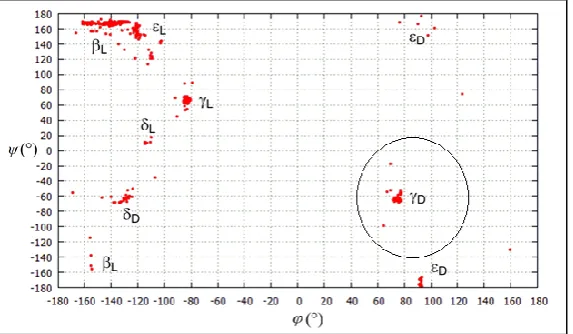
[image:2.595.156.443.211.378.2]a function of four independent variables, , χ1 and χ2 as defined in scheme 1. The Ramachandran map E = E (,) obtained using AM1/GA-MNC is represented in figure 1.
Figure 1: Representation of the Ramachandran map E(,) for N-For-Cys-NH2 obtained from AM1/GA-MNC calculation
Figure 1 shows all backbone conformers found for trans N-For-Cys-NH2 in isolated state. The backbone
conformation ɣD mentioned in figure 1 occupies 14% of the population of the last generation. The torsional
angles ≈ 73.8 and ≈ - 61.5° at AM1/GA-MNC calculation.
By associating the ɣD backbone conformation with the –CH2-SH side-chain conformation, six side-chain
conformers were located on the side-chain PES, E ɣD (1, 2) as shown in figure 2.
Figure 2: The optimized geometries of side-chain conformers found for D conformationslocated at AM1/AG-MNC calculation. In
brackets the relative energies calculated compared to the global minimum L [g+, g+] conformation having ΔHf = - 81.3 kcal/mol
The D backbone conformation is stabilized by intramolecular H-bonding C=O…H-N, which is characteristic to
[image:2.595.177.420.478.686.2]178
The optimized torsional angles, relative energies and intramolecular interactions for ɣD conformation of
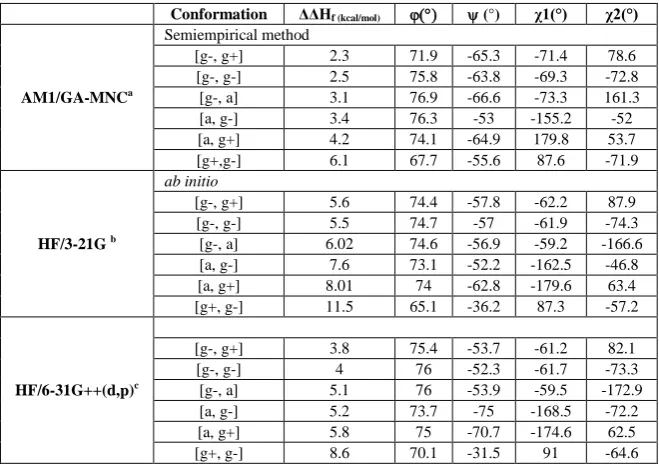
[image:3.595.133.463.132.364.2]N-For-L-Cys-NH2 are summarized in table 1 and table 2, respectively.
Table 1: Relative energies, bonds angles for ɣD conformations of N-For-L-Cys-NH2 optimized at AM1/GA-MNC, HF/3-21G and
HF/6-31G++(d, p) levels of theory
Conformation ΔΔHf (kcal/mol) (°) χ1(°) χ2(°)
AM1/GA-MNCa
Semiempirical method
[g-, g+] 2.3 71.9 -65.3 -71.4 78.6 [g-, g-] 2.5 75.8 -63.8 -69.3 -72.8 [g-, a] 3.1 76.9 -66.6 -73.3 161.3 [a, g-] 3.4 76.3 -53 -155.2 -52 [a, g+] 4.2 74.1 -64.9 179.8 53.7 [g+,g-] 6.1 67.7 -55.6 87.6 -71.9
HF/3-21G b
ab initio
[g-, g+] 5.6 74.4 -57.8 -62.2 87.9 [g-, g-] 5.5 74.7 -57 -61.9 -74.3 [g-, a] 6.02 74.6 -56.9 -59.2 -166.6 [a, g-] 7.6 73.1 -52.2 -162.5 -46.8 [a, g+] 8.01 74 -62.8 -179.6 63.4 [g+, g-] 11.5 65.1 -36.2 87.3 -57.2
HF/6-31G++(d,p)c
[g-, g+] 3.8 75.4 -53.7 -61.2 82.1 [g-, g-] 4 76 -52.3 -61.7 -73.3 [g-, a] 5.1 76 -53.9 -59.5 -172.9 [a, g-] 5.2 73.7 -75 -168.5 -72.2 [a, g+] 5.8 75 -70.7 -174.6 62.5 [g+, g-] 8.6 70.1 -31.5 91 -64.6
Relative energies were calculated compared to the global minimum L [g+, g+] conformation having: a ΔHf = - 81.3 kcal/mol; b ΔHf = -
808.03285866 hartrees; c ΔH
f =- 812.33016178 hartrees
Table 2: Summary of intramolecular interactions (in Å) in ɣD conformations of N-For-L-Cys-NH2 calculated at HF/3-21G and
HF/6-31G++(d, p) levels of theory
Conformation O7…H17S
O6…H9
(C7) O6…H17S S…H9 N4…H17S
HF/3-21G
[g-,g+] 1.93 3.12
[g-, g-] 1.9
[g-, a] 1.9
[a, g-] 2.55 1.89
[a, g+] 1.95
[g+,g-] 1.93 2.2
HF/6-31G++(d,p)
[g-, g+] 2.08
[g-, g-] 2.04
[g-, a] 2.05
[a, g-] 2.13
[a, g+] 2.15
[g+, g-] 2.14 2.31
From the data presented in tables 1 and 2, two of the six conformers (ɣD [a, g-] and ɣD [g+, g]) are stabilized by
SH…OCNH interaction, using the AM1/GA-MNC and the HF/3-21G calculation. While at the HF/6-31G++(d, p), one conformation (ɣD [g+, g-]) exhibit this interaction.
The energy gap between ɣD [g-, g+] and ɣD [g-, g-] (≈ 0.2 kcal/mol) is in relationship with the weak interaction
SH…NH, which is established in ɣD [g-, g+].
The geometrical form adopted by ɣD[g-, a] is similar to that of ɣD[g-, g-] with one exception. The position of the
acid hydrogen of sulfur as shown by the angle values χ1 in table 1. Thus, in ɣD[g-, a] the side chain –CH2-SH
becomes more close to the C7 which increases the steric effect in this conformation. Therefore, its heat of formation has increased by 0.6 kcal/mol.
The least stable minimum is ɣD[g+, g-]. According to AM1/GA-MNC results, both S…HNH and SH…OC side
chain-backbone interactions are favored for this conformation. While, only SH…OC is favored at HF/3-21G and HF/6-31G++(d, p) levels of theory. In addition, this conformation is more compact than the five others. For CH3CO-Cys-NHCH3 [8] and For-L-Ser-NH2 [5], all nine side chain conformers were located at the
179
Table 3: Relative energies and torsional angles of ɣD side-chain conformers found for For-L-Ser-NH2 [5] and CH3CO-Cys-NHCH3
[8] at the HF/3-21G
Conformation ΔΔHf (kcal/mol) (°) χ1(°) χ2(°)
For-L-Ser-NH2 [5]
[g-, g+] 12.5 72.2 -57.5 -61.1 79.1 [g-, g-] 12.9 74.7 -55.2 -57.6 -78.2 [g-, a] 12 75.4 -56.1 -58.6 176 [a, g-] 10.6 67.5 -31.2 -163.8 -39.9 [a, g+] 12 71.3 -52.2 170.8 49 [g+, g-] 9.4 78 -45.2 81.9 -62.2 [g+, g+] 14 62.9 -40.3 41.7 48.7 [g+, a] 17.1 51.9 -28.7 65.8 173.3
[a, a] 12.7 74 -65 -177.5 -158 CH3CO-Cys-NHCH3 [8]
[g-, g+] 5.7 75.7 -58.5 -60.8 88.9 [g-, g-] 5.54 76.3 -57.3 -60.6 -73.2 [g-, a] 6.04 76 -57.2 -58 -167.3 [a, g-] 7.55 73.6 -69.3 -174.1 -74.7 [a, g+] 8.26 74.7 -63.9 -178.8 62.5 [g+, g-] 11.7 66.8 -37.2 89 -57.6 [g+, g+] 12.45 47.2 -25.9 58.3 55.6 [g+, a] 13.66 55.6 -28.4 70.6 162.9
[a, a] 9.53 74.9 -62.1 -171.4 -176.6
For the two diamides models (For-L-Ser-NH2 and CH3CO-Cys-NHCH3), all nine side-chain conformers have
been found at the HF/3-21 level of theory. As can be seen from this table, three conformers (ɣD [g+, g+], ɣD [g+,
a] and ɣD [a, a]) are not located in our study using AM1/GA-MNC. Furthermore, the size difference between
sulfur and oxygen could play an important role in kinds of intramolecular interactions established and the energy quantities noticed.
At HF/3-21G, the relative energies and torsional angles values of side chain conformers ɣD[g-, g+], ɣD [g-, g-],
ɣD[g-, a], ɣD[a, g-], ɣD[a, g+] and ɣD[g+, g-] obtained for CH3CO-Cys-NHCH3 [8]and For-Cys-NH2 (this study)
are equal as can be seen from tables 1 and 3. Therefore, the AM1/GA-MNC provides optimized geometries that can be used as starting structures for ab initio calculation.
CONCLUSION
We carried out a conformational study on the preferred minima D (C7ax)of trans N-For-L-Cys-NH2 in isolated
state by AM1/GA-MNC calculation. All conformations located were subjected to geometry optimization at the HF/3-21G and the HF/6-31G++ (d, p) levels of theory. Six of the nine side-chain conformers were located ɣD[g-,
g+], ɣD [g-, g-], ɣD[g-, a], ɣD[a, g-], ɣD[a, g+] and ɣD[g+, g-].
The three levels of theory reported here (AM1/GA-MNC, HF/3-21G and HF/6-31G++(d,p)), Provide the same results. No conformational migrations were observed.
According to our calculations, the AM1/GA-MNC provides optimized geometries that can be used as starting structures for ab initio calculation. It can be a reliable technique for the study of conformational spaces of molecules with large sizes like proteins.
REFERENCES
[1] A Perczel; A K Fuzery; A G Csaszar. J. Comput. Chem., 2003,(24),1157-1171. [2] A Perczel; R Daudel; J Angyan; I G Csizmadia. Can J. Chem., 1990, (68), 1882-1888.
[3] O Farkas; A Perczel; JF Marcoccia; M Hollosi; IG Csizmadia. J. Mol. Struct. (Theochem), 1995, 331, 27-36.
[4] A Perczel; O Farkas; IG Csizmadia. J. Comput. Chem, 1996, 17, 821-834. [5] A Perczel; O Farkas; IG Csizmadia. J. Am. Chem. Soc., 1996,118, 7809-7817.
[6] JA Bombasaro; MA Zamora; HA Baldoni; RD Enriz. J. Phys. Chem., 2005, 109, 874-884.
[7] MA Zamora; HA Baldoni; AM Rodriguez; RD Enriz; CP Sosa; A Perczel; A Kucsman; O Farkas; E Deretey; JC Vank; IG Csizmadia. Can. J. Chem., 2002, 80, 832-844.
[8] MA Zamora; HA Baldoni; JA Bombasaro; ML Mak; A Perczel; O Farkas; RD Enriz. J. Mol. Struct. (Theochem)., 2001, 540, 271-283.
180
[10] MJ Frisch; GW Trucks; HB Schlegel; GE Scuseria; M ARobb; JR Cheeseman; G Scalmani; V Barone; B Mennucci; GA Petersson; H Nakatsuji; M Caricato; X Li; H P Hratchian; A F Izmaylov; J Bloino; G Zheng; JL Sonnenberg; M Hada; M Ehara; K Toyota; R Fukuda; J Hasegawa; M Ishida T Nakajima; Y Honda; O Kitao; H Nakai; T Vreven; J A Montgomery; J J E Peralta; F Ogliaro; M Bearpark; JJ Heyd E Brothers; KN Kudin; V N Staroverov; R Kobayashi; J Normand; K Raghavachari; A Rendell; JC Burant; S S Iyengar; J Tomasi; M Cossi; N Rega; JM Millam; M Klene; JE Knox; JB Cross; V Bakken; C Adamo; J Jaramillo; R Gomperts; RE Stratmann; O Yazyev; A J Austin; R Cammi; C comelli, J W Ochterski, R L Martin, K Morokuma, VG Zakrzewski, G A Voth, P Salvador, JJ Dannenberg, S Dapprich, A D Daniels, O Farkas, JB Foresman, JV Ortiz, J Cioslowski, DJ Fox, Gaussian, Inc., Wallingford CT, 2009.
![Table 3: Relative energies and torsional angles of ɣD side-chain conformers found for For-L-Ser-NH2 [5] and CH3CO-Cys-NHCH3 [8] at the HF/3-21G](https://thumb-us.123doks.com/thumbv2/123dok_us/8737412.889042/4.595.170.427.97.322/table-relative-energies-torsional-angles-chain-conformers-nhch.webp)
