0095-1137/07/$08.00
⫹
0
doi:10.1128/JCM.02317-06
Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Use of Outer Surface Protein Repeat Regions for Improved
Genotyping of
Staphylococcus epidermidis
䌤
Alastair B. Monk and Gordon L. Archer*
Division of Infectious Diseases, Department of Internal Medicine, Virginia Commonwealth University School of
Medicine, Richmond, Virginia 23298
Received 15 November 2006/Returned for modification 11 December 2006/Accepted 19 December 2006
Staphylococcus epidermidis
is an important nosocomial pathogen, but little is known of its epidemiology.
Accurate, reproducible typing systems would greatly improve epidemiologic investigations of
S. epidermidis
. The
sequence-based typing technique most recently evaluated, multilocus sequence typing (MLST), often lacks
discrimination and can be expensive. PCR and sequence-based analyses of the serine-aspartate repeat region
of
sdrG
(Fbe) and the repeat region of the accumulation-associated protein gene (
aap
) were evaluated for the
ability to discriminate among previously well-characterized
S
.
epidermidis
clinical isolates. Forty-eight strains
were investigated, with
sdrG
found in 100% and
aap
found in 79% of all strains tested. Both genes demonstrated
PCR product size and nucleotide sequence variation. Each system by itself gave an index of discrimination
similar in value to that of MLST (0.924 and 0.953 compared to 0.96), but discrimination was further improved
when combinations of the three systems were used. We conclude that typing systems using amino acid and
nucleotide repeat regions of the
S. epidermidis
surface proteins SdrG and Aap show promise as typing tools and
should be investigated using a larger panel of clinically relevant isolates.
Staphylococcus epidermidis
is one of the most important
causes of nosocomial bacteremia (26).
S
.
epidermidis
is
respon-sible for 50 to 70% of catheter-related and other foreign body
infections (26). The prerequisite for
S
.
epidermidis
foreign
body infections is biofilm formation, mediated initially by a
range of outer surface proteins, including staphylococcal
sur-face proteins (SSP1 and -2), autolysin proteins (AtlE), and an
accumulation-associated protein (Aap) (26). Biofilm
forma-tion can also occur via direct interacforma-tion between host
extra-cellular matrix proteins, such as fibrinogen, fibronectin, and
thrombospondin, and
S
.
epidermidis
outer surface proteins,
such as fibrinogen binding protein (Fbe). Fbe (
sdrG
) has
pre-viously been shown to promote adhesion to fibrinogen (3, 6)
and is a member of a family of serine-aspartate repeat proteins
expressed by
S
.
epidermidis
(14). There are three members of
the cell surface-associated serine-aspartate family of proteins
in
S
.
epidermidis
, namely, SdrF, SdrG (Fbe), and SdrH, and
they are all characterized by the distinctive serine-aspartate
dipeptide (SD) repeats (14). SdrG (Fbe) is a 93.7-kDa protein
with a 50-residue signal sequence proximal to the amino
ter-minus and serine-aspartate repeats proximal to the LPXTG
motif at the carboxy terminus (2, 14). Accumulation-associated
protein (Aap) has previously been shown to be associated with
biofilm formation and adherence to cells (19, 22). Aap has also
been shown to be a poor marker of whether an
S. epidermidis
isolate is commensal or invasive (20, 25), although it is
consid-ered a virulence factor (20). Aap is a surface-exposed outer
surface protein of 140 kDa composed of an N-terminal domain
of short, 16-amino-acid (aa) repeats, 13 repeats of 128 aa, and
19 repeats of 6 aa, with an LPXTG and transmembrane
do-main at the C terminus (2).
Little is known of the epidemiology of
S
.
epidermidis
in the
health care or carriage setting, and the basis for
S
.
epidermidis
epidemiology has mostly been pulsed-field gel electrophoresis
(PFGE) analysis (12, 23). PFGE has been shown to be highly
discriminatory but is labor-intensive and costly, and problems
exist with standardization for interlaboratory reproducibility
(15). Other typing systems in current use for
S
.
epidermidis
include randomly amplified polymorphic DNA analysis (13),
multilocus variable number of tandem repeat analysis (8), and
amplified fragment length polymorphism analysis (21). All of
these systems are also gel-based band typing systems, and
problems exist with the reproducibility of patterns between
gels and labs. Multilocus sequencing typing (MLST) is a
well-established method for discriminating among bacterial isolates
that utilizes the DNA sequences of internal fragments of
so-called housekeeping genes in order to assign strains to
se-quence types (STs) (28). This system is highly reproducible
since it is based on sequence data, but it may not distinguish
among individual isolates. Two different MLST systems have
previously been reported for
S
.
epidermidis
(27, 28), but the
recent publication of a condensed MLST system combining
elements of all three systems to give the most discriminatory
scheme is the one that was used in this investigation. For
Staphylococcus aureus
, the addition of a typing system based on
the sequence of the repeat region in surface protein A
(en-coded by
spa
) is also reproducible and adds a greater level of
discrimination to MLST (17).
In the present work, the nucleotide serine-aspartate repeat
regions of
sdrG
(Fbe) and the nucleotides of
aap
encoding the
six-amino-acid repeat region were amplified via PCR and
se-quenced from a wide range of previously well-characterized
isolates of
S
.
epidermidis
. The repeat regions were analyzed to
see if they could provide increased discrimination to MLST
* Corresponding author. Mailing address: Virginia Commonwealth
University School of Medicine, Sanger Hall, Room 1-018, 1101 East
Marshall St., Richmond, VA 23298. Phone: (804) 828-0673. Fax: (804)
828-5022. E-mail: [email protected].
䌤
Published ahead of print on 3 January 2007.
730
on May 16, 2020 by guest
http://jcm.asm.org/
and be used to independently type
S
.
epidermidis.
We chose to
compare only sequence-based rather than gel-based (e.g.,
PFGE or randomly amplified polymorphic DNA analysis)
typ-ing systems because we felt that they were more reproducible
and could more easily be applied for general use. It has also
been shown that MLST is equally as discriminatory as PFGE
for the closely related species
S
.
aureus
(16).
MATERIALS AND METHODS
Bacterial strains and media.A selection of 48 isolates previously well char-acterized via SCCmectyping and MLST (28) were used to assess whethersdrG
(Fbe) repeats andaaprepeats can discriminate among isolates ofS.epidermidis. The 48 isolates were composed of 30 isolates collected from the blood of patients with prosthetic valve endocarditis (PVE) and 17 isolates from the blood of patients without PVE collected as part of the Surveillance and Control of Patho-gens of Epidemiological Importance (SCOPE) study (4). A total of 28 PVE isolates were methicillin resistant, and 2 were methicillin susceptible, while 10 of the SCOPE isolates were methicillin resistant and 7 were methicillin susceptible. The final isolate (RP62A28) was an isolate (RP62A) repeatedly passaged for 28 days in order to test the in vitro stability of the repeat regions. All strains were grown in brain heart infusion broth or agar (Becton Dickinson, Sparks, MD) at 37°C, with shaking at 220 rpm.
MLST typing.Genomic DNA was extracted using a QIAGEN DNA miniprep kit (QIAGEN, Hilden, Germany). Seven genes were used for this MLST system, as recently described by Thomas et al. (24). Primers (24) were designed to amplify carbamate kinase (arcC), shikimate dehydrogenase (aroE), glutamyl-tRNA reductase (gtr), pyrimidine biosynthesis (pyr), DNA mismatch repair pro-tein (mutS), triosephosphate isomerase (tpi), and acetyl coenzyme A acetyltrans-ferase (yqiL) genes. PCR amplification of the genes was performed in 50-l reaction mixes composed of 1l of template DNA, 1l of each primer set (100 pmol of each primer), 18l of sterile distilled water, and 30l of QIAGEN PCR Supermix (QIAGEN, Hilden, Germany). The thermocycler conditions were as follows: 95°C for 2 min and then 35 cycles of 95°C for 30 seconds, 55°C for 30 seconds, and extension at 72°C for 2 min for all genes. The genes were visualized
on a 1.5% acrylamide gel stained with ethidium bromide. All PCRs were cleaned up using a QiaQuick PCR purification kit (QIAGEN, Valencia, CA). Nucleotide sequences were obtained for PCR products in both directions, using the same primer sets as those for amplification (at 10 pmol), with an annealing temperature of 55°C and with BigDye fluorescent terminators on an ABI Prism 3700 instrument. Contigs of sequence data were constructed using Vector NTI v7.1 (Invitrogen, Carlsbad, CA), using maximum stringency, and were edited manually. Alleles were assigned in comparison to the MLST website (http://www.mlst.net).
PCR amplification of repeat regions.Genomic DNA was extracted using a QIAGEN DNA miniprep kit (QIAGEN, Hilden, Germany). The size of thesdrG
repeat region was established using PCR primers SDTYPING1F (5⬘CTCAGA AGGCAATTCTGTATGG 3⬘) and SDTYPING1R (5⬘AACGCTCCTAAACC TGCAAA 3⬘), and theaaprepeat region size was established using PCR primers AAPREPEATF (5⬘ TCACTAAACAACCTGTTGACGAA 3⬘) and AAPRE PEATR (5⬘AATTGATTTTTATTATCTGTTGAATGC 3⬘). Both sets of prim-ers were designed using the RP62A genome. Amplification was performed in 50-l reaction mixes composed of 1l of template DNA, 1l of each primer (100 pmol of each), 18l of sterile distilled water, and 30l of QIAGEN PCR Supermix (QIAGEN, Hilden, Germany). The thermocycler conditions were as follows: 95°C for 2 min and then 35 cycles of 95°C for 30 seconds, 55°C for 30 seconds, and extension at 72°C for 2 min for both genes. The genes were visualized on a 1.5% acrylamide gel stained with ethidium bromide.
Repeat region sequencing.All PCRs were cleaned up using a QiaQuick PCR purification kit (QIAGEN, Valencia, CA). Nucleotide sequences were obtained for PCR products in both directions forsdrG, using SDTYPING1R and SDTYPING2F (5⬘CAACAACAACTGATGAAAATGGA 3⬘), and foraap, using AAPREPEATF and AAPREPEATR, using the primers at 10 pmol, an annealing temperature of 55°C, and BigDye fluorescent terminators, on an ABI Prism 3700 instrument. Con-tigs of sequence data were constructed using Vector NTI v7.1 (Invitrogen, Carlsbad, CA), using maximum stringency, and were edited manually. Once assembled, the nucleotide data were cross checked for PCR amplicon size.
Assignment of alleles and sequence types.The nucleotide coding regions for the SD repeat region of thesdrGgene product and the six-amino-acid repeat region of theaapgene product (Fig. 1) were used to discriminate among isolates. Initially, the described repeat regions of RP62A were used to assign alleles, as shown in Fig. 1, based upon differences in sequence and size. The nucleotide
FIG. 1. Genes studied in this investigation. The genes are shown schematically, with the repeat regions of interest as well as primer binding sites.
*
, PCR and sequencing primer binding sites (arrows show an alternative sequencing primer for
sdrG
only); S, signal peptide; A, ligand binding
domain; B, repeat region; SD, serine-aspartate repeats; W, cell wall spanning domain. Repeat regions are shown in bold italics. (Adapted from
Microbiology
[2] with permission of the publisher.)
on May 16, 2020 by guest
http://jcm.asm.org/
sequences of the repeat regions from the remaining isolates were then identified by comparison to RP62A, and individual 12-, 18-, or 21-base-pair repeats were used to generate a repeat region database. Any unique unidentified repeats were assigned a number, and the sequence of individual repeats or numbers described the ST for that isolate. The numbers were assigned in sequence as each differing repeat was encountered, beginning with RP62A. Thus, the RP62AsdrGrepeat region had a pattern of 1-2-3-4-5-6-7-8-9, indicating that there were nine sets of 18- or 21-base-pair repeats in this region; this pattern described sequence type 1.
[image:3.585.47.542.78.621.2]Sequence type 2 became 1-10-2-11-7-4-2-12-8, and so on (Table 1). A similar system was followed for assigning repeat numbers toaap(Table 1). Lineages were assigned from the SD typing nucleotide data on the basis of global clus-tering via the use of Clustal W (1). The clusclus-tering grouped the repeat regions into subsets of sequence data that were arbitrarily defined as lineages on the basis of minor rearrangements and point mutations between the members of the lineage and major rearrangements and region differences between members of different lineages. Although the initial sequence types were assigned by visual inspection,
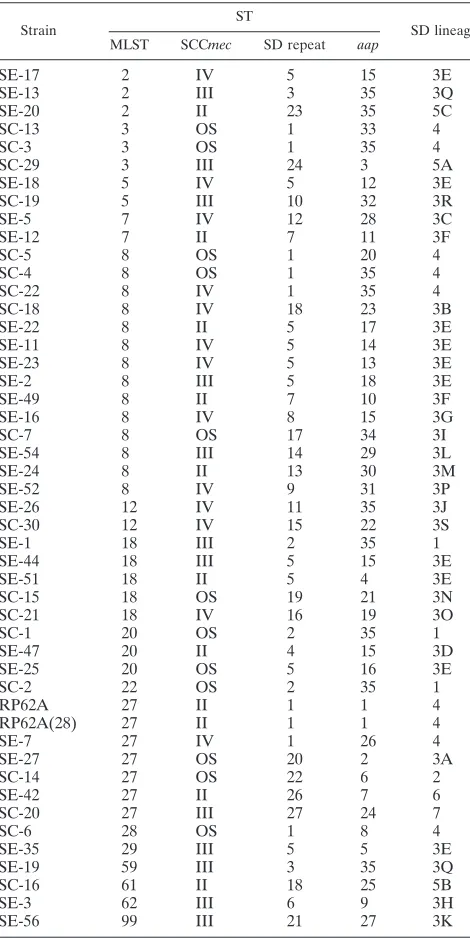
TABLE 1. Examples of repeat regions for Aap STs and SD repeat STs in this investigation
ST Lineage Repeat
Aap STs
1 1-1-2-3-2-3-1-1-1-2-4-3-2-3-2-3-3-5 2 1-1-1-2-1-2-3-1-1-1-2-3-1-2-3-6-2-3-1 3 1-1-1-3-1-3-1-3-1-1-1-1-1-1-3-5-6-7 4 1-1-2-1-9-10-1-1-1-2-3-2-3-6-2-3-1-7-8
5 1-11-12-3-13-3-1-1-1-14-15-3-14-3-2-3-5-2-16-1-17-18-4-17-17-3-19-5-20-21 6 1-2-3-14-3-2-22-3-1-1-2-23-3-14-3-17-3-24-3-5-6
7 25-26-3-2-4-3-1-1-14-29-3-14-3-2-3-14-3-5-6 8 30-1-2-31-32-10-33-1-17-22-3-14-3-34-3-17-3-5
9 35-35-36-35
10 1-1-1-2-3-1-1-1-1-2-3-1-1-2-3-2-3-2-37-38 11 1-1-1-2-3-1-1-1-1-2-3-1-1-9-3-2-3-2 12 1-1-2-1-2-3-1-1-1-1-2-3-1-2-3-6-1 13 1-1-2-1-2-3-1-1-1-17-1-1-2-3-6-2-3-1-7 14 1-1-2-1-2-3-1-1-1-17-3-1-2-3-6-2-3-1-7 15 1-1-2-1-2-3-1-1-1-2-3-1-2-3-6-2-3-1 15 1-1-2-1-2-3-1-1-1-2-3-1-2-3-6-2-3-1 15 1-1-2-1-2-3-1-1-1-2-3-1-2-3-6-2-3-1 15 1-1-2-1-2-3-1-1-1-2-3-1-2-3-6-2-3-1 16 1-1-2-1-2-3-1-7
17 1-1-2-1-2-1-1-1-1-17-1-1-17-3-6-1-7
18 1-1-2-3
19 1-1-2-3-17-10-2-3-3-1-1-17-3-17-3-5-6-7 20 1-1-2-3-17-11-1-1-2-23-3-2-3-2-3-5-6-7 21 1-1-2-3-17-11-14-1-1-1-1-17-3-17-3-5-6-7 22 1-1-2-3-2-10-2-3-3-1-1-2-3-2-3-5 23 1-1-2-3-2-3-2-3-1-1-1-1-2-3-5-6 24 1-1-14-3-2-3-2-3-1-1-1-2-3-2-3-5
25 12-11-14-3-14-3-1-1-2-4-3-2-3-2-3-2-3-5-6-7 26 1-2-1-2-3-1-1-1-2-3-1-2-3-6-2-3-1 27 1-1-1-2-3-2-22-3-1-2-4-3-14-3-2-3-5 28 1-2-3-2-3-1-1-1-2-4-3-2-3-2-3-5 29 1-2-3-2-3-2-4-3-1-1-2-23-3-2-3-2-3-2-3-5 30 1-2-3-2-3-2-4-3-1-1-2-4-3-2-3-2-3-2-3-5-6 31 14-1-17-3-1-1-1-17-3-1-14-3-6-2-3-1 32 1-2-3-2-4-3-1-1-2-4-3-2-3-2-3-2-3-5 33 1-1-1-1-2-3-1
34 1-1-1-4-3-2-3-3-1--2-3-1-3-1-1-1-2-3
35 Not present
SD repeat STs
1 4 1-2-3-4-5-6-7-8-9 2 1 1-10-5-11-3-4-5-12-9
3 3Q 13-14-15-16-17-18-19-20-13-14-14-21-20-14-20-14-15-14-21-20-15-34-15-41
4 3D 13-14-15-16-14-17-18-19-20-14-15-14-21-20-14-15-14-15-14-21-20-14-15-14-21-20-15-22-23-21-24-15-14-21-20-15-41 5 3E 13-14-15-16-14-17-18-19-20-14-15-14-21-20-14-15-14-15-14-21-20-15-22-23-21-24-15-14-21--20-15-41
6 3H 13-14-15-16-14-17-18-19-20-14-15-14-21-20-14-15-14-15-14-21-15-22-23-21-24-15-41 7 3F 13-14-15-16-14-17-18-19-20-14-15-14-21-20-14-15-14-15-14-21-20-15-22-23-21-24-15-14-21-41 8 3F 13-14-15-16-14-17-18-19-20-14-15-14-21-20-14-15-14-15-14-21-20-15-22-23-21-24-15-14-21-41 9 3G 13-14-15-16-14-17-18-19-20-14-15-14-21-20-14-15-14-15-14-21-20-15-22-23-24-15-14-21-20-15-41 10 3P 13-14-15-16-17-18-19-20-14-15-14-21-20-14-15-14-15-14-21-20-15-22-23-21-24-15-14-21-20-15-41 11 3R 13-14-15-16-17-20-14-18-14-19-20-14-15-14-21-20-14-15-14-15-14-21-20-15-22-23-21-24-15 12 3J 13-14-15-16-14-17-20-14-25-26-27-28-29-30-31-28-32-30-33
13 3C 13-14-15-16-14-17-17-20-14-25-26-27-28-29-30-31-28-32-30-33
14 3M 13-14-15-16-14-17-20-17-16-17-18-19-20-14-15-14-21-20-14-15-14-15-14-21-20-15-22-23-21-24-15-14-15-14-15-20-15-34-35-41 15 3L
13-14-15-16-14-17-20-17-16-17-18-19-20-14-15-14-21-20-14-15-14-15-14-21-20-15-22-23-21-24-15-14-15-14-15-14-21-20-15-34-35-41
16 3S 13-14-25-26-29-36-37-14-21-26-14-15-14-15-13-21-20-15-22-26-23-21-34-15-41
17 3I 13-14-15-16-14-17-18-19-20-14-25-26-29-36-37-14-21-20-14-15-14-15-39-21-20-15-22-26-23-21-34-15 18 3O 13-14-15-16-14-17-68-18-19-20-14-25-26-29-36-37-14-38-20-13-15-14-15-39-21-20-15-22-26-23-21-34-15-41 19 3B 13-14-15-16-14-18-16-18-19-20-14-25-26-29-36-37-14-38-20-14-15-14-15-39-21-20-15-22-40-23-21-34-15-41 20 3N 13-14-15-16-14-17-68-18-19-20-14-25-26-29-40-37-14-21-20-14-15-14-15-39-21-20-15-22-23-21-34-15
21 3A 13-14-15-16-14-17-16-17-18-19-20-14-15-14-21-20-14-15-14-15-14-21-20-15-22-23-21-24-15-14-15-14-65-20-15-34-58-59 22 3K 13-14-15-16-14-17-20-17-16-17-18-69-20-14-15-14-21-20-14-15-14-15-14-21-20-15-22-23-21-24-15-14-60-14-21-20-15-34 22 2 13-30-15-30-41-2-41-2-41-5-41-32-46-2-31-32-46-47-32
23 5C 43-44-5-54-39-36-30-31-5-54-52-29-29-54-37-50-31-53-5-3-30-10-27-5-36-28-11-29-37-41-28-11-31-55-56-28-11-28-57-45 24 5A 43-44-5-54-344-36-29-31-5-54-38-40-38-66-36-29-36-5-31-28-29-36-5-31-5-31-67-29-5-54-27-28-29-30-31-28-32-30-33 25 5B 43-44-5-54-38-40-29-31-5-54-48-29-29-54-49-50-31-28-3-30-10-27-5-36-28-11-29-37-36-28-11-31-55-56-28-11-46-58 26 6 61-62-42-16-14-17-45-17-18-17-18-19-20-14-15-14-21-22-14-20-14-15-14-21-20-15-22-23-21-24-15-14-15-14-21-20-15-34-35-41 27 7 63-64-36-11-36-11-36-5-10-5-11-51
on May 16, 2020 by guest
http://jcm.asm.org/
a computer program assigned new repeat numbers and designated the sequence types for isolates typed later.
Statistical analysis.Synonymous and nonsynonymous mutations and ratios were calculated using the Nei-Gojobori (Jukes-Cantor-corrected) method, with pairwise deletion handling of gaps; the standard error was determined using 1,000 bootstrap replicates.Ztests for neutrality of mutations as well as for synonymous and nonsynonymous mutations were all calculated using MEGA v3.1 (11). Discrimination of the typing systems was calculated using Simpson’s index of diversity (7), which indicates the probability that two random isolates from a population will have different genotypes.
RESULTS
Isolate information.
MLST divided the 48 strains into 16
STs, with the predominant STs being ST8 (
n
⫽
14) and ST27
(
n
⫽
7) and with ST2, -3, and -20 each composed of three
isolates. ST5 and -7 were composed of two isolates each, and
ST22, -28, -29, -59, -61, -62, and -99 were all composed of single
isolates, as shown in Table 2. SD typing divided the isolates
into 27 STs, and
aap
typing divided the isolates into 35 STs. SD
ST5 was the predominant SD ST (
n
⫽
9), with ST1 (
n
⫽
3) and
ST7 (
n
⫽
2) being the next most prevalent sequence types. All
other sequence types were composed of single isolates only.
Aap ST35 (
n
⫽
9) was the predominant Aap ST, and ST15
(
n
⫽
4) was the next most predominant ST, with all other STs
composed of single isolates only. SD typing also divided the
isolates into five major lineages, with lineages 3 and 5 sharing
the majority of isolates, as shown in Table 2. Only two isolates
shared an identical MLST, SD typing, and Aap typing repeat,
and these were genotypically MLST ST8, SD ST5, and Aap
ST35.
sdrG
SD repeat types, sizes, and lineages.
sdrG
was chosen
as the initial gene to be analyzed, as other single genes with
sufficient repeat regions did not display as much variation (A.
Monk, unpublished data). Serine-aspartate repeats have
pre-viously been shown to allow a high degree of discrimination in
S. aureus
(10). Initial surveys revealed the largest amount of
size variation in
sdrG
PCR amplicons, and the gene was
present in all strains surveyed (14). There were three
differ-ently sized PCR amplicons of the SD repeat region from the 48
strains analyzed (
⬃
200 bp,
⬃
4 to 500 bp, and
⬃
8 to 900 bp),
and there was 100% concordance between the size of the PCR
fragment and the number of repeat cassettes. The DNA
se-quence revealed 69 alleles of the repeat cassette, composed of
1 21-bp, 4 12-bp, and 64 different 18-bp repeats. These
com-bined to give 27 different STs and seven lineages, as shown in
Table 1. The SD typing system had a Simpson’s index of
dis-crimination of 0.924, which is less than the MLST value of 0.96,
as shown in Table 3. However, SD typing managed to
subdi-vide identical MLST types. Forty-one isolates in this
investiga-tion fell into only nine MLST types (STs 2, 3, 5, 7, 8, 12, 18, 20,
and 27). When SD typing and MLST were used in conjunction,
the 41 isolates fell into 32 different STs overall, and the index
of discrimination increased to 0.992, as shown in Table 3. In
addition, identical STs of the SD typing system are present in
multiple different MLST backgrounds, which suggests that
there may have been genetic exchange and recombination of
sdrG
between different genetic backgrounds of
S
.
epidermidis
.
aap
repeats, sizes, and types.
The
aap
repeat region was
detected via PCR in 38 of the 48 strains in this study (79%).
[image:4.585.44.279.89.558.2]aap
was used in this investigation as an additional typing
sys-tem because even though it was not present in all isolates, the
TABLE 2. MLST, SCC
mec
, SD repeat, and
aap
typing STs for
each isolate in this investigation
Strain
ST
SD lineage MLST SCCmec SD repeat aap
SE-17
2
IV
5
15
3E
SE-13
2
III
3
35
3Q
SE-20
2
II
23
35
5C
SC-13
3
OS
1
33
4
SC-3
3
OS
1
35
4
SC-29
3
III
24
3
5A
SE-18
5
IV
5
12
3E
SC-19
5
III
10
32
3R
SE-5
7
IV
12
28
3C
SE-12
7
II
7
11
3F
SC-5
8
OS
1
20
4
SC-4
8
OS
1
35
4
SC-22
8
IV
1
35
4
SC-18
8
IV
18
23
3B
SE-22
8
II
5
17
3E
SE-11
8
IV
5
14
3E
SE-23
8
IV
5
13
3E
SE-2
8
III
5
18
3E
SE-49
8
II
7
10
3F
SE-16
8
IV
8
15
3G
SC-7
8
OS
17
34
3I
SE-54
8
III
14
29
3L
SE-24
8
II
13
30
3M
SE-52
8
IV
9
31
3P
SE-26
12
IV
11
35
3J
SC-30
12
IV
15
22
3S
SE-1
18
III
2
35
1
SE-44
18
III
5
15
3E
SE-51
18
II
5
4
3E
SC-15
18
OS
19
21
3N
SC-21
18
IV
16
19
3O
SC-1
20
OS
2
35
1
SE-47
20
II
4
15
3D
SE-25
20
OS
5
16
3E
SC-2
22
OS
2
35
1
RP62A
27
II
1
1
4
RP62A(28)
27
II
1
1
4
SE-7
27
IV
1
26
4
SE-27
27
OS
20
2
3A
SC-14
27
OS
22
6
2
SE-42
27
II
26
7
6
SC-20
27
III
27
24
7
SC-6
28
OS
1
8
4
SE-35
29
III
5
5
3E
SE-19
59
III
3
35
3Q
SC-16
61
II
18
25
5B
SE-3
62
III
6
9
3H
[image:4.585.301.542.90.182.2]SE-56
99
III
21
27
3K
TABLE 3. Simpson’s indexes of diversity for typing systems
considered in this investigation
Typing system Index of discrimination (D)
MLST
0.96
SD typing
0.924
aap
typing
0.954
MLST plus SD typing
0.992
SD typing plus
aap
typing
0.994
MLST plus
aap
typing
0.996
MLST plus SD typing plus
aap
typing
0.997
on May 16, 2020 by guest
http://jcm.asm.org/
gene exhibited high variability in both PCR and sequencing
assays. In addition,
aap
has some similarity to
spa
of
S
.
aureus
,
a proven epidemiological marker. There were three differently
sized PCR amplicons of the
aap
repeat region (data not
shown), with 100% concordance between the size of the PCR
fragment and the number of repeat cassettes. Thirty-six alleles
of the
aap
repeat cassette were composed of 38 different
18-base-pair repeats and combined to give 35 different STs, as
shown in Table 1. The
aap
typing system had a Simpson’s index
of discrimination of 0.954, which is almost equal to that of the
MLST system alone (0.96). When combined with MLST,
aap
typing increased the index of diversity to 0.997 (Table 3), with
the 41 isolates previously mentioned being subdivided into 38
different STs.
Evolutionary pressure on both repeat regions.
The SD
re-peat region cassettes always started with TC, and the overall
repeat was structured as follows: TC(X
1)(X
2)(X
3)(X
4)(X
5)
(X
1)(X
1)(X
6)(X
3)(X
7)(X
8)(X
2)(X
1)(X
3)(X
2)(X
4), where X
1is
A, G, C, or T, X
2is A, G, or C, X
3is A or G, X
4is T or C, X
5is T or A, X
6is T or G, X
7is T, C, or A, and X
8is A or T. The
SD repeat region had a synonymous/nonsynonymous mutation
(
dS
/
dN
) ratio of 4.2, indicating that the SD repeat region is
under the influence of purifying selection. A result of
⬍
1
would have indicated that the region is under the influence of
positive selection, 1 would indicate no selection at all, and a
result of
⬎
1 indicates purifying selection. Purifying selection is
thought to have occurred when existing amino acids are
se-lected to stay the same by pressure against nonsynonymous
mutations which would change an amino acid. The
dS
value
was 0.89 (standard error, 0.17), and the
dN
value was 0.24
(standard error, 0.17). A
Z
test for purifying selection was
highly significant (
P
⫽
0.00014). While mutation would appear
to be the main source of variation within the SD repeat alleles,
slip-strand mispairing has commonly been observed in
clfB
(an
SD gene with similarity to
sdrG
) and
spa
, which have repeat
regions used for typing of
S
.
aureus
(9, 10).
The 18-bp repeat of
aap
had a highly conserved repeat pattern
of CC(X
1)(X
2)(X
3)(X
4)(X
5)(X
6)(X
6)(X
6)(X
7)(X
1)(X
4)(X
6)(X
3)
(X
4)(X
6)(X
4), where X
1is A or T, X
2is G or A, X
3is C, T, A,
or G, X
4is G, A, or T, X
5is G, C, or A, X
6is A, T, or C, and
X
7is C or A, and had a
dS
/
dN
ratio of 0.24, indicating that the
repeat region is under the influence of positive selection. A
Z
test for positive selection was performed and was also
signifi-cant (
P
⫽
0.00031). Both repeat regions also appeared to be
stable, since in vitro passaging of RP62A for 28 days on brain
heart infusion agar did not change the profile of either repeat.
DISCUSSION
Standardized, highly discriminatory, reproducible typing
sys-tems are needed for distinguishing among
S
.
epidermidis
iso-lates because of their importance as nosocomial pathogens.
Highly variable genes encoding outer surface proteins that
interact with the environment or the host have been shown to
be as informative for typing as housekeeping genes for the
closely related species
S
.
aureus
(18). In this paper, we describe
two new nucleotide sequence-based typing systems that utilize
only single gene products and have high discrimination (equal
to or slightly less than that of MLST). The two gene systems
described in this paper also have the ability to increase the
resolution of MLST data, as these systems could be used to
further subdivide identical MLST STs. The genes chosen have
regions of repeated sequence, and both the sequence of the
repeats and the number of repeats vary among isolates.
How-ever, the repeat regions appear to be stable when isolates are
passaged in vitro, making these regions good candidates for
epidemiological typing markers.
We have shown possible evidence of recombination in the
SD repeat region of
sdrG
, with identical SD types being found
in multiple previously defined STs and lineages. The
recombi-nation noted in the SD repeats of
sdrG
explains the lower
discrimination for this typing system than that for MLST and
Aap typing due to shared alleles, with STs present in multiple
backgrounds. It also suggests that
S
.
epidermidis
undergoes
genome diversification by interstrain genetic exchange and
re-combination. This is supported by recent work where
meta-bolic genes called “housekeeping” genes in the MLST system
have been shown to be four times more likely to diversify via
recombination than by mutation in
S
.
epidermidis
(M. C.
En-right, personal communication). Recombination can mask
phylogenetic relationships, as previously shown (5). The
re-combination discovered in
sdrG
of
S. epidermidis
is similar to
that noted in the SD repeat region of
clfB
from the closely
related organism
S
.
aureus
(10).
The two gene systems studied in this paper are under
dif-ferent selection pressures. The SD typing region is under
strong purifying selection pressure, suggesting that the
charac-teristic SD amino acid repeats are under pressure to stay the
same, producing a stable evolutionary marker, although it is
not as discriminatory as Aap or the targets of MLST. In
con-trast, the
aap
region is under positive selection, perhaps due to
its previously noted function as a virulence factor. The high
rate of variation at this gene locus is an important
character-istic of genes that are useful as epidemiological markers. We
have shown that the
aap
gene can be used as an independent
epidemiological marker, with equal discrimination to MLST
(0.954 and 0.96, respectively). However,
aap
is not present in
every strain, and another study found the gene to be present in
88% of colonizing and 68% of invasive isolates (20), similar to
the value found in our study (79%). Yet the presence or
ab-sence of
aap
by itself may be a genotypic marker with both
typing and clinical implications. This possibility requires
addi-tional investigation with a larger set of isolates that have been
characterized clinically. We have also demonstrated that either
single-gene epidemiological marker can impart higher
discrim-ination to the MLST system (index of discrimdiscrim-ination, 0.994 for
MLST plus SD typing, 0.996 for MLST plus
aap
typing, and
0.96 for MLST alone), and both together are also better than
MLST alone (0.994 for SD plus
aap
typing versus 0.96 for
MLST). Thus, the use of single-gene versus multiple-gene
(e.g., MLST) typing systems should be driven by cost (single
genes would be cheaper), the need for more discrimination
(localized epidemiology for the more discriminatory gene
re-peat systems, as opposed to large-scale evolutionary studies for
MLST), and time. We suggest using
aap
typing as a simple,
inexpensive epidemiological genotyping method while using
MLST in conjunction with SD and
aap
typing for long-term
evolutionary studies.
on May 16, 2020 by guest
http://jcm.asm.org/
ACKNOWLEDGMENT
This work was supported by grant 5R01AI35705-13 from the
Na-tional Institute of Allergy and Infectious Diseases.
REFERENCES
1.Aiyar, A.2000. The use of CLUSTAL W and CLUSTAL X for multiple sequence alignment. Methods Mol. Biol.132:221–241.
2.Bowden, M. G., W. Chen, J. Singvall, Y. Xu, S. J. Peacock, V. Valtulina, P. Speziale, and M. Hook.2005. Identification and preliminary characterization of cell-wall-anchored proteins ofStaphylococcus epidermidis. Microbiology
151:1453–1464.
3.Davis, S. L., S. Gurusiddappa, K. W. McCrea, S. Perkins, and M. Hook.
2001. SdrG, a fibrinogen-binding bacterial adhesin of the microbial surface components recognizing adhesive matrix molecules subfamily from Staphy-lococcus epidermidis, targets the thrombin cleavage site in the Bbeta chain. J. Biol. Chem.276:27799–27805.
4.Edmond, M. B., S. E. Wallace, D. K. McClish, M. A. Pfaller, R. N. Jones, and R. P. Wenzel.1999. Nosocomial bloodstream infections in United States hospitals: a three-year analysis. Clin. Infect. Dis.29:239–244.
5.Feil, E. J., E. C. Holmes, D. E. Bessen, M. S. Chan, N. P. Day, M. C. Enright, R. Goldstein, D. W. Hood, A. Kalia, C. E. Moore, J. Zhou, and B. G. Spratt.
2001. Recombination within natural populations of pathogenic bacteria: short-term empirical estimates and long-term phylogenetic consequences. Proc. Natl. Acad. Sci. USA98:182–187.
6.Hartford, O., L. O’Brien, K. Schofield, J. Wells, and T. J. Foster.2001. The Fbe (SdrG) protein ofStaphylococcus epidermidisHB promotes bacterial adherence to fibrinogen. Microbiology147:2545–2552.
7.Hunter, P. R., and M. A. Gaston.1988. Numerical index of the discrimina-tory ability of typing systems: an application of Simpson’s index of diversity. J. Clin. Microbiol.26:2465–2466.
8.Johansson, A., S. Koskiniemi, P. Gottfridsson, J. Wistrom, and T. Monsen.
2006. Multiple-locus variable-number tandem repeat analysis for typing of
Staphylococcus epidermidis. J. Clin. Microbiol.44:260–265.
9.Koreen, L., S. V. Ramaswamy, E. A. Graviss, S. Naidich, J. M. Musser, and B. N. Kreiswirth.2004.spatyping method for discriminating among Staph-ylococcus aureusisolates: implications for use of a single marker to detect genetic micro- and macrovariation. J. Clin. Microbiol.42:792–799. 10.Koreen, L., S. V. Ramaswamy, S. Naidich, I. V. Koreen, G. R. Graff, E. A.
Graviss, and B. N. Kreiswirth.2005. Comparative sequencing of the serine-aspartate repeat-encoding region of the clumping factor B gene (clfB) for resolution within clonal groups ofStaphylococcus aureus. J. Clin. Microbiol.
43:3985–3994.
11.Kumar, S., K. Tamura, and M. Nei.2004. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform.5:150–163.
12.Linhardt, F., W. Ziebuhr, P. Meyer, W. Witte, and J. Hacker.1992. Pulsed-field gel electrophoresis of genomic restriction fragments as a tool for the epidemiological analysis ofStaphylococcus aureus and coagulase-negative staphylococci. FEMS Microbiol. Lett.74:181–185.
13.Marquet-Van Der Mee, N., S. Mallet, J. Loulergue, and A. Audurier.1995. Typing ofStaphylococcus epidermidis strains by random amplification of polymorphic DNA. FEMS Microbiol. Lett.128:39–44.
14.McCrea, K. W., O. Hartford, S. Davis, D. N. Eidhin, G. Lina, P. Speziale, T. J. Foster, and M. Hook.2000. The serine-aspartate repeat (Sdr) protein family inStaphylococcus epidermidis. Microbiology146:1535–1546. 15.Murchan, S., M. E. Kaufmann, A. Deplano, R. de Ryck, M. Struelens, C. E.
Zinn, V. Fussing, S. Salmenlinna, J. Vuopio-Varkila, N. El Solh, C. Cuny, W. Witte, P. T. Tassios, N. Legakis, W. van Leeuwen, A. van Belkum, A. Vindel, I. Laconcha, J. Garaizar, S. Haeggman, B. Olsson-Liljequist, U. Ransjo, G. Coombes, and B. Cookson.2003. Harmonization of pulsed-field gel electro-phoresis protocols for epidemiological typing of strains of methicillin-resis-tantStaphylococcus aureus: a single approach developed by consensus in 10 European laboratories and its application for tracing the spread of related strains. J. Clin. Microbiol.41:1574–1585.
16.Peacock, S. J., G. D. de Silva, A. Justice, A. Cowland, C. E. Moore, C. G. Winearls, and N. P. Day.2002. Comparison of multilocus sequence typing and pulsed-field gel electrophoresis as tools for typingStaphylococcus aureus isolates in a microepidemiological setting. J. Clin. Microbiol.40:3764–3770. 17.Robinson, D. A., and M. C. Enright.2003. Evolutionary models of the emergence of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother.47:3926–3934.
18.Robinson, D. A., A. B. Monk, J. E. Cooper, E. J. Feil, and M. C. Enright.
2005. Evolutionary genetics of the accessory gene regulator (agr) locus in
Staphylococcus aureus. J. Bacteriol.187:8312–8321.
19.Rohde, H., C. Burdelski, K. Bartscht, M. Hussain, F. Buck, M. A. Horstkotte, J. K. Knobloch, C. Heilmann, M. Herrmann, and D. Mack.
2005. Induction ofStaphylococcus epidermidisbiofilm formation via proteo-lytic processing of the accumulation-associated protein by staphylococcal and host proteases. Mol. Microbiol.55:1883–1895.
20.Rohde, H., M. Kalitzky, N. Kroger, S. Scherpe, M. A. Horstkotte, J. K. Knobloch, A. R. Zander, and D. Mack.2004. Detection of virulence-associ-ated genes not useful for discriminating between invasive and commensal
Staphylococcus epidermidis strains from a bone marrow transplant unit. J. Clin. Microbiol.42:5614–5619.
21.Sloos, J. H., P. Janssen, C. P. van Boven, and L. Dijkshoorn.1998. AFLP typing ofStaphylococcus epidermidisin multiple sequential blood cultures. Res. Microbiol.149:221–228.
22.Sun, D., M. A. Accavitti, and J. D. Bryers.2005. Inhibition of biofilm for-mation by monoclonal antibodies againstStaphylococcus epidermidisRP62A accumulation-associated protein. Clin. Diagn. Lab. Immunol.12:93–100. 23.Tenover, F. C., R. D. Arbeit, R. V. Goering, P. A. Mickelsen, B. E. Murray,
D. H. Persing, and B. Swaminathan.1995. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J. Clin. Microbiol.33:2233–2239.
24.Thomas, J. C., M. R. Vargas, M. Miragaia, S. J. Peacock, G. L. Archer, and M. C. Enright.6 December 2006. An improved multilocus sequence typing scheme forStaphylococcus epidermidis. J. Clin. Microbiol. doi:10.1128/JCM. 01934-06.
25.Vandecasteele, S. J., W. E. Peetermans, R. Merckx, B. J. Rijnders, and J. Van Eldere.2003. Reliability of the ica, aap and atlE genes in the discrim-ination between invasive, colonizing and contaminantStaphylococcus epider-midisisolates in the diagnosis of catheter-related infections. Clin. Microbiol. Infect.9:114–119.
26.von Eiff, C., G. Peters, and C. Heilmann.2002. Pathogenesis of infections due to coagulase-negative staphylococci. Lancet Infect. Dis.2:677–685. 27.Wang, X. M., L. Noble, B. N. Kreiswirth, W. Eisner, W. McClements, K. U.
Jansen, and A. S. Anderson.2003. Evaluation of a multilocus sequence typing system forStaphylococcus epidermidis. J. Med. Microbiol.52:989–998. 28.Wisplinghoff, H., A. E. Rosato, M. C. Enright, M. Noto, W. Craig, and G. L. Archer. 2003. Related clones containing SCCmec type IV predominate among clinically significantStaphylococcus epidermidisisolates. Antimicrob. Agents Chemother.47:3574–3579.