FORMULATION DEVELOPMENT AND EVALUATION OF SOLID
LIPID NANOPARTICLES OF BUPRENORPHINE
Suryakalyanam Vikas*, S. Poonguzhali, Manohar Babu S.
Department of Industrial Pharmacy, SIMS College of Pharmacy, SIMS Group of Institutions,
Mangaldas Nagar, Guntur,-522001, Andhra Pradesh, India.
ABSTRACT
Buprenorphine is a poorly water soluble drug as it is a BCS Class II
drug and the oral bioavailability is very less due to the extensive first
pass effect. So there is a need to develop a formulation that can
improve the oral bioavailability. In this work, efforts were made to
prepare stable solid lipid nanoparticles for enhancement of oral
bioavailability of Buprenorphine. Drug solubility studies were
performed with various lipids to test the solubility and the glyceryl
monostearate was the lipid in which the drug is more soluble. SLNs
were prepared by the hot homogenization method. For the optimization
of the surfactant the three more formulations were prepared.
Characterization of SLNs was done by measuring particle size, Poly
dispersity index and zeta potential by zeta sizer. The Entrapment
efficiency of the SLNs was found to be in the range of 80 to 95% In- vitro drug release
studies were performed for all these SLNs for 24 hrs in pH 6.8 phosphate buffer. In these
studies the cumulative percentage drug release from all these formulations showed prolonged
release. The formulation (F2) was found to be promising prolonged release. This formulation
(F2) released 77.3 % of drug in 24 hours. The release kinetics of the optimized formulation is
first order and from the higuchi plot the release is by diffusion process and from the
korsemeyers plot the diffusion process is non-fickian diffusion.
KEYWORDS: Buprenorphine, Solid Lipid Nanoparticles, Bioavailability, Glyceryl
monostearate, Solubility studies.
Volume 5, Issue 9, 1387-1397. Research Article ISSN 2277– 7105
*Corresponding Author
Suryakalyanam Vikas
Department of Industrial
Pharmacy, SIMS College of
Pharmacy, SIMS Group of
Institutions, Mangaldas
Nagar, Guntur,-522001,
Andhra Pradesh, India. Article Received on 10 July 2016,
Revised on 01 Aug. 2016, Accepted on 22 Aug. 2016
1. INTRODUCTION
Novel Drug Delivery Systems: Novel Drug Delivery Systems (NDDS) has captured the
interest of both national and international pharmaceutical research organizations. Pioneering
research to devise newer strategies for effectively delivering the drugs in the body or
improvise the existing technologies to enhance their efficiency is the need of the hour. Since
inception, NDDS has seen a foray of transformations right from microencapsulation,
epithelial and transdermal delivery, liposomal vesicles and nanoparticles. The ever improving
delivery systems are not only beneficial to the patients as they are less cumbersome and easy
to abide by but also reduce the complications associated with the induction of the drug in the
body. These novel strategies will effectively potentiate drug administration ensuring safe and
effective therapeutic regimen.[1-4]
The NDDS essentially consists of the drug against the causative agent of the disease being
treated and a carrier system into which the drug is loaded and transported to site of action.
Efforts are now being made to devise carriers that can transport multiple drugs and release
them on command.[5]
Nanoparticulate system
Colloidal particles ranging in size between 10 and 1000 nm are known as nanoparticles. They
are manufactured from synthetic/natural polymers and ideally suited to optimize drug
delivery and reduce toxicity. Over the years, they have emerged as a variable substitute to
liposomes as drug carriers. The successful implementation of nanoparticles for drug delivery
depends on their ability to penetrate through several anatomical barriers, sustained release of
their contents and their stability in the nanometer size. However, the scarcity of safe polymers
with regulatory approval and their high cost have limited the wide spread application of
nanoparticles to clinical medicine.[6, 7]
To overcome these limitations of polymeric nanoparticles, lipids have been put forward as an
alternative carrier, particularly for lipophilic pharmaceuticals. These lipid nanoparticles are
known as solid lipid nanoparticles (SLNs), which are attracting wide attention of formulators
world-wide.[8]
SLNs are colloidal carriers developed in the last decade as an alternative system to the
existing traditional carriers (emulsions, liposomes and polymeric nanoparticles). They are a
substituted by a solid lipid. SLN offer unique properties such as small size, large surface area,
high drug loading and the interaction of phases at the interfaces, and are attractive for their
potential to improve performance of pharmaceuticals, neutraceuticals and other materials.[6-8]
Solid lipid nanoparticles (SLN) are a class of particulate drug carriers made from lipids that
remain in the solid state at room and body temperatures. Lipids utilized for SLN are typically
physiological lipids, including: fatty acids, steroids, waxes, mono-, di-, or triglyceride
mixtures. A wide variety of biocompatible surfactants are used to stabilize SLN; therefore,
SLN have the advantage of physical stability with low toxicity. SLN are becoming
increasingly used for the protection of labile drugs from degradation in the body and for
sustained release.[8]
2. MATERIALS AND METHODS
2.1. Materials used
S. No MATERIAL
1 Buprenorphine
2 Glycerol Monostearate 3 Precirol
4 Stearic Acid 5 Oleic Acid 6 Dyanasan 114 7 Dyanasan 118 8 Compritol ATO 888 9 Tween 80
10 Di-sodium hydrogen phosphate 11 Potassium di-hydrogen phosphate
12 Acetone
13 Iso Propyl Alcohol
2.2. Methods Used
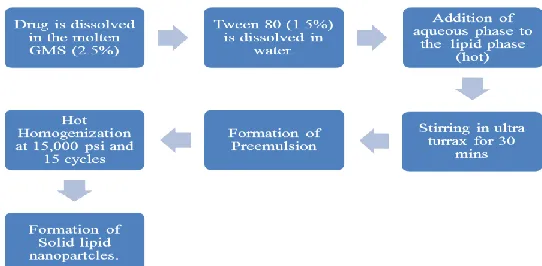
[image:3.596.161.432.611.744.2]Schematic representation of optimizad formulation of solid lipid nanoparticles.
3. RESULTS AND DISCUSSION
Table No-1: Composition of the formulation
Formulation code
Lipids (%)
Drug (mg) Tween 80 (%)
GMS Precirol Dyanasan
F1 2.5 - - 10 1.5
F2 5 - - 10 1.5
F3 7.5 - - 10 1.5
F4 - 2.5 - 10 1.5
F5 - 5 - 10 1.5
F6 - - 2.5 10 1.5
F7 - - 5 10 1.5
F8 5 - - 10 0.5
F9 5 - - 10 1
F10 5 - - 10 2
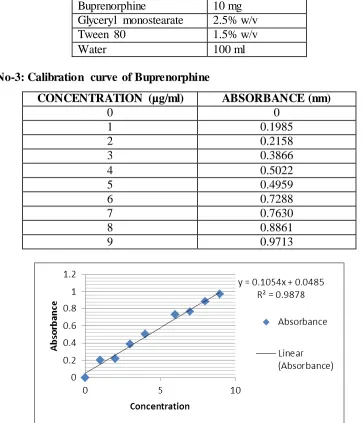
Table No-2: Optimized Formulation
Buprenorphine 10 mg
Glyceryl monostearate 2.5% w/v
Tween 80 1.5% w/v
Water 100 ml
Table No-3: Calibration curve of Buprenorphine
CONCENTRATION (µg/ml) ABSORBANCE (nm)
0 0
1 0.1985
2 0.2158
3 0.3866
4 0.5022
5 0.4959
6 0.7288
7 0.7630
8 0.8861
9 0.9713
Table No-4: Solubility studies of the drug in various lipids
S No Lipid Qty Drug Qty Solubility
1 Compritol ATO 1 g
10 mg
Insoluble
2 Precirol 0.35 g Soluble
3 Glyceryl monostearate 0.3 g Soluble
4 Dyanasan 118 0.5 g Soluble
5 Dyanasan 114 0.4 g Soluble
Table No-5: Particle size distribution of Solvent Evaporation method
S. No Surfactant Concentration ( w/v) Size PdI
1 0.5% 1579 1.000
2 1% 993.9 0.309
3 2% 611.3 0.881
Table No-6: Optimization of the lipid concentration
S. No Lipid (%w/v) Size (nm) PDI
1 2.5 241.5 1
2 5 247.55 0.998
3 7.5 773.85 1
Table No-7: Optimization of the surfactant concentration
S. No Tween 80 concentration (% w/v) Size (nm) PDI
F8 0.5 520 0.999
F9 1 400 0.998
F2 1.5 280 0.772
F10 2 350 0.998
Table No-8: Zeta potential data after the Homogenization.
Formulation Zeta potential
F1 -23.9±1.87
F2 -22.7±1.78
F3 -28.1±2.16
F4 -26.3±2.06
F5 -19.9±1.56
F6 -18.5±1.45
F7 -18.1±1.35
Table No-9: Entrapment Efficiency data
Formulation Entrapment efficiency (%)
F1 91.2
F2 95.01
F3 85.68
F4 89.23
F5 90.08
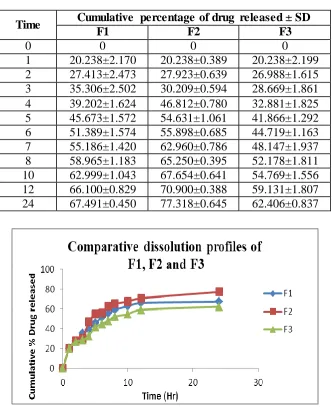
Table No-10: Comparative dissolution data of SLNs of F1, F2 and F3
Table No-11: Comparative dissolution data of SLNs of F4, F5 and F6
Fig No-3: Comparative dissolution profiles of F1, F2 and F3
Time Cumulative % of drug released ± SD
F4 F5 F6
0 0 0 0
1 18.537±1.206 20.238±2.199 15.476±0.390
2 20.934±1.710 26.988±1.615 16.821±0.740
3 22.827±1.047 28.669±1.861 20.621±0.524
4 24.886±1.216 41.866±1.292 23.296±0.649
5 27.543±1.564 44.719±1.163 28.122±0.537
6 30.460±1.674 48.147±1.937 31.656±1.026
7 33.975±1.437 52.178±1.811 36.198±0.652
8 38.432±1.750 54.769±1.556 39.389±1.175
10 44.003±1.857 59.131±1.807 43.928±1.043
12 48.480±2.080 62.406±0.837 47.119±1.179
24 55.242±2.082 65.329±0.974 50.552±1.043
Time Cumulative percentage of drug released ± SD
F1 F2 F3
0 0 0 0
1 20.238±2.170 20.238±0.389 20.238±2.199
2 27.413±2.473 27.923±0.639 26.988±1.615
3 35.306±2.502 30.209±0.594 28.669±1.861
4 39.202±1.624 46.812±0.780 32.881±1.825
5 45.673±1.572 54.631±1.061 41.866±1.292
6 51.389±1.574 55.898±0.685 44.719±1.163
7 55.186±1.420 62.960±0.786 48.147±1.937
8 58.965±1.183 65.250±0.395 52.178±1.811
10 62.999±1.043 67.654±0.641 54.769±1.556
12 66.100±0.829 70.900±0.388 59.131±1.807
Fig No-4: Comparative dissolution profiles of F4, F5 and F6.
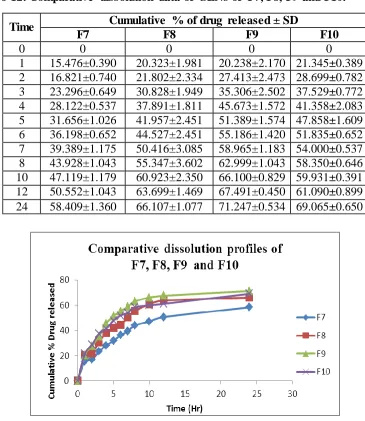
Table No-12: Comparative dissolution data of SLNs of F7, F8, F9 and F10.
Fig No-5: Comparative dissolution profiles of F7, F8, F9 and F10
Time Cumulative % of drug released ± SD
F7 F8 F9 F10
0 0 0 0 0
[image:7.596.113.478.288.709.2].
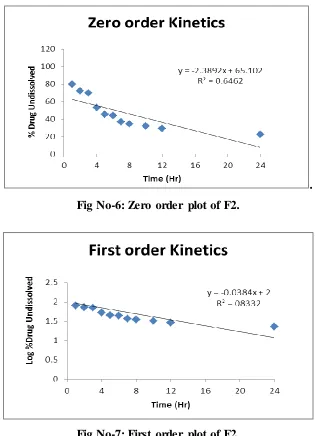
Fig No-6: Zero order plot of F2.
[image:8.596.142.452.73.254.2]Fig No-7: First order plot of F2.
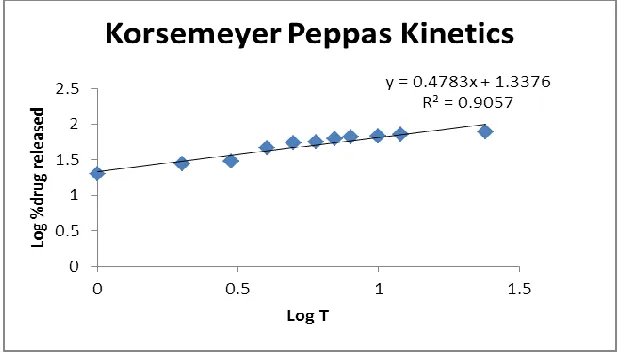
[image:8.596.140.457.486.718.2]Fig No-9: Korsemeyer peppas plot of F2
Table No-13: Evaluation data of optimized formulation (F2)
Parameters Data observed
Particle size 56.94
Zeta potential -22.7±1.78
Entrapment efficiency 95.01
In vitro dissolution release 77.31
Zero order plot R2=0.646
First order plot R2=0.8332
Higuchi plot R2=0.9152
[image:9.596.143.452.73.253.2]Korsemeyer peppas plot R2=0.905
Table No-14: Stability Studies at room temperature and refrigerated temperature
Day At room temperature At refrigerated temperature
Size(nm) PDI Zeta potential (mV) Size(nm) PDI Zeta potential (mV)
1 174.73±4.473 0.305±0.097 -28.1±2.16 194.73±4.473 0.405±0.097 -28.1±2.16
30 205.56±8.58 0.309±0.146 -26.3±2.06 294.23±2.898 0.410±0.012 -26.1±2.04
60 210.9±11.49 0.308±0.066 -26.4±2.08 300.0±8.54 0.411±0.031 -26.66±2.12
4. CONCLUSION
Buprenorphine is a poorly water soluble drug as it is a BCS Class II drug and the oral
bioavailability is very less due to the extensive first pass effect. so there is a need to develop a
formulation that can improve the oral bioavailability .In this work, efforts were made to
prepare stable solid lipid nanoparticles for enhancement of oral bioavailability of
Buprenorphine. Drug solubility studies were performed with various lipids to test the
solubility and the glyceryl monostearate was the lipid in which the drug is more soluble.
The preparation of the solid lipid nanoparticles was initially done by solvent evaporation
As the organic solvents are also used in the above process so the method is changed to the hot
homogenization process. Initially, all the process parameters were optimized for the
homogenization time and cycles.
SLNs were prepared by the hot homogenization method. All the seven SLNs were prepared
with the three lipids with each lipid at least two formulations and the formulations with the
optimized lipid is three. For the optimization of the surfactant the three more formulations
were prepared. Characterization of SLNs was done by measuring particle size, Poly dispersity
index and zeta potential by zeta sizer. The Entrapment efficiency of the SLNs was found to
be in the range of 80 to 95% In-vitro drug release studies were performed for all these SLNs
for 24 hrs in pH 6.8 phosphate buffer. In these studies the cumulative percentage drug release
from all these formulations showed prolonged release. The formulation (F2) was found to be
promising prolonged release. This formulation (F2) released 77.3 % of drug in 24 hours. The
release kinetics of the optimized formulation is first order and from the higuchi plot the
release is by diffusion process and from the korsemeyers plot the diffusion process is
non-fickian diffusion. Stability studies were conducted for finally optimized formulation (F2) at
room temperature and 4oC for 2 months; no appreciable changes were noticed in size and zeta
potential.
5. BIBILIOGRAPHY
1. Allen DD, Lockman PR, Oyewumi MO, Koziara JM, Roder KE, Mumper RJ., Brain
uptake of thiamine-coated nanoparticles, J.Control. Release 2003; 93: 271–282.
2. Bargoni A, Cavalli R, Zara GP., Transmucosal transport of tobramycin incorporated in
solid lipid nanoparticles (SLN) after duodenal administration to rats. Part II – Tissue
distribution. Pharm Res. 2001; 43: 497–502.
3. Bodkin JA, Zornberg GL, Lukas SE, Cole JO: Buprenorphine treatment of refractory
depression. J Clin Psychopharmacol. Feb., 1995; 15(1): 49-57.
4. B. Siekmann, K. Westesen., Sub-micron sized parenteral carrier systems based on solid
lipid, Pharm. Pharmacol. Lett. 1992; 1: 123-126.
5. Cavalli R, Gasco MR, Chetoni P., Solid lipid nanoparticles (SLN) as ocular delivery
system for tobramycin. Int J Pharm. 2002; 238: 241–5.
6. Chen H, Chang X, Du D., Podophyllotoxin-loaded solid lipid nanoparticles for epidermal
targeting. J Control Release. 2006; 110: 296–306.
7. C. Freitas C, Müller RH., Effect of light and temperature on zeta potential and physical
8. C.M. O'Driscoll, B.T. Griffin; Biopharmaceutical challenges associated with drugs with
low aqueous solubility— The potential impact of lipid-based formulations; Advanced