organic papers
o966
Krugeret al. C28H30O3 doi:10.1107/S160053680600403X Acta Cryst.(2006). E62, o966–o968
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
A pentacycloundecane dimer
Hendrik G. Kruger, Melanie Rademeyer* and Reshika Ramdhani
School of Chemistry, Howard College Campus, University of KwaZulu–Natal, Durban 4041, South Africa
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 173 K
Mean(C–C) = 0.002 A˚ Disorder in main residue Rfactor = 0.042 wRfactor = 0.132
Data-to-parameter ratio = 17.4
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 8 November 2005 Accepted 2 February 2006
#2006 International Union of Crystallography All rights reserved
The title compound, 6,6-(3,6-diprop-2-enylpentacyclo-[6.2.1.02 , 7.04 , 1 0.05 , 9 ]undecane-3,6-diyldioxy)pentacyclo-[6.2.1.02,7.04,10.05,9]undecan-3-one, C28H30O3, is a
pentacyclo-undecane dimer. The molecule is chiral and contains two polycyclic pentacycloundecane cage units. Both enantiomers are present in the crystal structure. In the molecule, certain C—C bond lengths deviate from the normal value, with both short and long bonds present.
Comment
The structure of the pentacycloundecane (PCU) dimer, (I), was determined as part of an ongoing study of the chemical reactivity and solid-state structures of substituted polycyclic PCU cage compounds.
The Grignard reaction with pentacycloundecane dione was first reported by Marchand et al.(2001). Nucleophilic attack on the PCU dione occurs almost exclusively from theexoface, producing an endo–endodiol as the main product. The cage dimer, (I), is formed when the cage dione is reacted with an equimolar amount of allyl magnesium bromide (1:1 instead of 4:1), but the mechanism for the formation of the cage dimer is not obvious.
A number of studies have focused on the molecular geometries of PCU cage derivatives, and it has been reported that these compounds exhibit C—C single bonds for which the bond lengths deviate from the expected value of 1.54 A˚ (Flippen-Andersonet al., 1991; Lindenet al., 2005; Krugeret al., 2005). These reports indicated that in a typical simple PCU cage derivative, the C9—C10 bond may be longer than normal, whereas some bonds involving atoms C4, C8 and C11 may be shorter than expected.
The cage skeleton has two faces, a ‘front side’ and a ‘back side’. The front side is where the C—O—C ether bridge can form and the back side is where the cyclobutane is positioned. In principle, two sets of diastereomers can form when the cage dimerizes, the starting material being amesocompound. One set can potentially dimerize at atom C1 with (a) the cyclo-butane rings of the two cages in a cisposition and (b) the cyclobutane rings in a trans position. The second set of diastereomeric dimers can potentially dimerize at atom C8 with the samecisandtransconfigurations. The mechanism of the dimerization reaction is not obvious and it is not clear why the two diastereomeric dimers do not form. In (I), the two cyclobutane rings on each cage skeleton are in acisposition. As observed for other PCU cage derivatives, the cage dimer exhibits C—C single bonds lengths (Table 1) that deviate from the expected value. The longest bonds observed in both cages are opposite to the cyclobutane ring [C9—C10 = 1.5866 (17) A˚ and C26—C27 = 1.5805 (19) A˚ ]. In both cages, the four bonds forming the cyclobutane rings are longer than expected (Table 1). The bonds involving the bridgehead atoms C4 and C21 are short [range 1.521 (2)–1.531 (2) A˚ ].
The unsymmetrical substitution at the mouth of cage two appears to affect the bond lengths in this cage; the bonds involving atom C25, at the ketone substituent, are shorter than normal [C24—C25 = 1.5185 (18) A˚ and C25—C26 = 1.514 (2) A˚ ]. In the related dione cage (Lindenet al., 2005), the corresponding bond lengths are also short, and range from 1.503 (4) to 1.517 (4) A˚ . The corresponding bonds on the opposite side of the second cage are not affected as severely, with C18—C28 falling within the expected range, and C27— C28 slightly shorter than normal. In the same way, C22—C23
and C22—C26 on the ketone-substituted side of the second cage are long, 1.563 (2) and 1.5585 (19) A˚ (also observed for the dione), but the C19—C20 and C20—C27 bonds on the other side of the cage do not deviate significantly from the expected value.
The five-membered rings defined by atoms C2–C6 and C3– C5/C9/C10 adopt envelope conformations, with C4 in the flap positions, and the rings C1–C3/C10/C11 and C5–C9 are twisted about C10—C11 and C8—C9, respectively. The five-membered rings defined by C19–C23 and C20–C22/C26/C27 in the second cage exhibit envelope conformations, with C21 in the flap positions, and the rings defined by C18–C20/C27/C28 and C22–C26 are twisted about C27—C28 and C25—C26, respectively.
The molecular packing is illustrated in Fig. 2. Molecules pack in layers parallel to the crystallographicbcplane.
Experimental
The synthesis of theendo–endocage diol is described by Marchandet al.(2001) and Govenderet al.(2003). When the ratio of Grignard reagent to dione is limited to 1:1, the dimer (I) is formed and may be recrystallized from an ethyl acetate–n-hexane (3:2) mixture.
Crystal data
C28H30O3
Mr= 414.52
Triclinic,P1
a= 6.1843 (17) A˚
b= 10.963 (3) A˚
c= 15.572 (4) A˚
= 86.327 (5)
= 83.046 (5)
= 80.221 (5)
V= 1031.7 (5) A˚3
Z= 2
Dx= 1.334 Mg m
3
MoKradiation Cell parameters from 990
reflections
= 3.4–28.3
= 0.09 mm1
T= 173 (2) K
Irregular fragment, colourless
0.510.440.30 mm
Data collection
Bruker SMART CCD area-detector diffractometer
’and!scans
Absorption correction: none 10401 measured reflections 4949 independent reflections
3894 reflections withI> 2(I)
Rint= 0.016 max= 28.0
h=8!7
k=14!14
l=20!20
organic papers
Acta Cryst.(2006). E62, o966–o968 Krugeret al. C
[image:2.610.47.297.71.323.2]28H30O3
o967
Figure 1
Molecular structure of (I) showing the atomic numbering scheme and displacement ellipsoids at the 50% probability level for non-H atoms (ORTEP-3; Farrugia, 1997).
Figure 2
[image:2.610.317.564.74.238.2]Refinement
Refinement onF2 R[F2> 2(F2)] = 0.042
wR(F2) = 0.132
S= 1.05 4949 reflections 284 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.0843P)2
+ 0.1152P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.37 e A˚
3
min=0.20 e A˚
[image:3.610.45.297.110.376.2]3
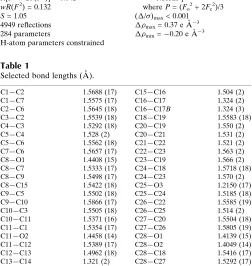
Table 1
Selected bond lengths (A˚ ).
C1—C2 1.5688 (17)
C1—C7 1.5575 (17)
C2—C6 1.5645 (18)
C3—C2 1.5539 (18)
C4—C3 1.5292 (18)
C5—C4 1.528 (2)
C5—C6 1.5562 (18)
C7—C6 1.5657 (17)
C8—O1 1.4408 (15)
C8—C7 1.5333 (17)
C8—C9 1.5498 (17)
C8—C15 1.5422 (18)
C9—C5 1.5502 (18)
C9—C10 1.5866 (17)
C10—C3 1.5505 (18)
C10—C11 1.5371 (16)
C11—C1 1.5354 (17)
C11—O2 1.4458 (14)
C11—C12 1.5389 (17)
C12—C13 1.4962 (18)
C13—C14 1.321 (2)
C15—C16 1.504 (2)
C16—C17 1.324 (2)
C16—C17B 1.324 (3)
C18—C19 1.5583 (18)
C20—C19 1.550 (2)
C20—C21 1.531 (2)
C21—C22 1.521 (2)
C22—C23 1.563 (2)
C23—C19 1.566 (2)
C24—C18 1.5718 (18)
C24—C23 1.570 (2)
C25—O3 1.2150 (17)
C25—C24 1.5185 (18)
C26—C22 1.5585 (19)
C26—C25 1.514 (2)
C27—C20 1.5504 (18)
C27—C26 1.5805 (19)
C28—O1 1.4139 (15)
C28—O2 1.4049 (14)
C28—C18 1.5416 (17)
C28—C27 1.5292 (17)
Atom C17, belonging to an allyl group, was found to be disordered and was refined over two positions, C17 and C17B, with C16—C17 and C16—C17Abond lengths restrained to be equal with an s.u. of 0.02 A˚ . Site occupancy factors were refined with their sum constrained to 1, and converged to 0.872 (4) for C17 and 0.128 (4) for C17B. Atom C17B, having a small contribution to the structure factors, was refined with an isotropic displacement parameter. All H
atoms were placed in calculated positions and refined using a riding model (C—H = 1.00 A˚ for methine CH, 0.99 A˚ for methylene CH2
and 0.95 A˚ for allyl CH). For all H atoms,Uiso(H) = 1.2Ueq(parent
atom).
Data collection: SMART-NT (Bruker, 1998); cell refinement:
SAINT-Plus(Bruker, 1999); data reduction:SAINT-Plus; program(s) used to solve structure:SHELXTL(Bruker, 1999); program(s) used to refine structure:SHELXL97(Sheldrick, 1997); molecular graphics:
MERCURY (Bruno et al., 2002) and ORTEP-3 for Windows
(Farrugia, 1997); software used to prepare material for publication:
WinGX(Farrugia, 1999) andPLATON(Spek, 2003).
The authors thank the Jan Boeyens Structural Chemistry Laboratory of the University of the Witwatersrand, South Africa, for the structure analysis.
References
Bruker (1998). SMART-NT. Version 5.050. Bruker AXS Inc., Madison,
Wisconsin, USA.
Bruker (1999). SAINT-Plus (Version 6.02) and SHELXTL (Version 5.1). Bruker AXS Inc., Madison, Wisconsin, USA.
Bruno, I. J., Cole, J. C., Edgington, P. R., Kessler, M. K., Macrae, C. F., McCabe, P., Pearson, J. & Taylor, R. (2002).Acta Cryst.B58, 389–397.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565. Farrugia, L. J. (1999).J. Appl. Cryst.32, 837–838.
Flippen-Anderson, J. L., George, C., Gilardi, R., Zajac, W. W., Walters, T. R., Marchand, A., Dave, P. R. & Arney, B. E. Jr (1991).Acta Cryst.C47, 813– 817.
Govender, T., Hariprakasha, H. K., Kruger, H. G. & Marchand, A. P. (2003).
Tetrahedron Asymmetry,14, 1553–1557.
Kruger, H. G., Rademeyer, M. & Ramdhani, R. (2005).Acta Cryst.E61, o3968–o3970.
Linden, A., Romanski, J., Mloston, G. & Heimgartner, H. (2005).Acta Cryst.
C61, o221–o226.
Marchand, A. P., Huang, Z., Chen, Z., Hariprakasha, H. K., Namboothiri, I. N. N., Brodbelt, J. S. & Reyzer, M. L. J. (2001).J. Heterocycl. Chem.38, 1361–1368.
Sheldrick, G. M. (1997).SHELXL97. University of Go¨ttingen, Germany. Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
organic papers
o968
Krugeret al. Csupporting information
sup-1
Acta Cryst. (2006). E62, o966–o968
supporting information
Acta Cryst. (2006). E62, o966–o968 [https://doi.org/10.1107/S160053680600403X]
A pentacycloundecane dimer
Hendrik G. Kruger, Melanie Rademeyer and Reshika Ramdhani
6,6-(3,6-diprop-2-enylpentacyclo[6.2.1.02,7.04,10.05,9]undecane-
3,6-diyldioxy)pentacyclo[6.2.1.02,7.04,10.05,9]undecan-3-one
Crystal data C28H30O3 Mr = 414.52
Triclinic, P1 Hall symbol: -P 1 a = 6.1843 (17) Å b = 10.963 (3) Å c = 15.572 (4) Å α = 86.327 (5)° β = 83.046 (5)° γ = 80.221 (5)° V = 1031.7 (5) Å3
Z = 2 F(000) = 444 Dx = 1.334 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 990 reflections θ = 3.4–28.3°
µ = 0.09 mm−1 T = 173 K
Irregular fragment, colourless 0.51 × 0.44 × 0.30 mm
Data collection
Bruker SMART CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
10401 measured reflections 4949 independent reflections
3894 reflections with I > 2σ(I) Rint = 0.016
θmax = 28.0°, θmin = 1.3° h = −8→7
k = −14→14 l = −20→20
Refinement Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.042 wR(F2) = 0.132 S = 1.05 4949 reflections 284 parameters 15 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0843P)2 + 0.1152P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.37 e Å−3 Δρmin = −0.20 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full
supporting information
sup-2
Acta Cryst. (2006). E62, o966–o968
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq Occ. (<1) O1 0.47664 (14) 0.77587 (8) 0.16772 (6) 0.0242 (2)
O2 0.36540 (14) 0.69130 (8) 0.30506 (5) 0.0214 (2) O3 0.24823 (18) 0.80195 (10) 0.49046 (6) 0.0369 (3) C1 0.7623 (2) 0.57241 (11) 0.28006 (8) 0.0211 (2) C2 0.8473 (2) 0.43742 (12) 0.24931 (8) 0.0243 (3) C3 0.6430 (2) 0.37796 (12) 0.23679 (9) 0.0257 (3) C4 0.6941 (2) 0.33215 (13) 0.14467 (9) 0.0305 (3) C5 0.7403 (2) 0.45626 (12) 0.10251 (9) 0.0275 (3) C6 0.9154 (2) 0.49109 (12) 0.15632 (8) 0.0254 (3) C7 0.8328 (2) 0.62564 (11) 0.18771 (8) 0.0222 (3) C8 0.6368 (2) 0.67266 (11) 0.13613 (8) 0.0220 (3) C9 0.5375 (2) 0.55218 (11) 0.13520 (8) 0.0231 (3) C10 0.4678 (2) 0.49621 (11) 0.22898 (8) 0.0221 (3) C11 0.51299 (19) 0.57390 (11) 0.30159 (8) 0.0198 (2) C12 0.4714 (2) 0.51335 (11) 0.39269 (8) 0.0228 (3) C13 0.2463 (2) 0.47902 (12) 0.41411 (8) 0.0263 (3) C14 0.2091 (3) 0.36548 (14) 0.43650 (9) 0.0347 (3) C15 0.7183 (2) 0.71987 (14) 0.04448 (9) 0.0306 (3) C16 0.8162 (2) 0.83617 (14) 0.04573 (9) 0.0354 (3)
C17 1.0303 (3) 0.83945 (18) 0.04105 (12) 0.0415 (5) 0.872 (4) C17B 0.7222 (14) 0.9473 (3) 0.0187 (9) 0.050* 0.128 (4) C18 0.6048 (2) 0.85400 (11) 0.29436 (8) 0.0229 (3)
supporting information
sup-3
Acta Cryst. (2006). E62, o966–o968
H9 0.4165 0.5612 0.0970 0.028* H10 0.3134 0.4780 0.2361 0.026* H12A 0.5828 0.4377 0.3980 0.027* H12B 0.4934 0.5712 0.4359 0.027* H13 0.1223 0.5429 0.4114 0.032* H14A 0.3296 0.2995 0.4398 0.042* H14B 0.0619 0.3499 0.4493 0.042* H15A 0.8309 0.6546 0.0168 0.037* H15B 0.5929 0.7362 0.0091 0.037*
H16A 0.7176 0.9126 0.0502 0.043* 0.872 (4) H16B 0.9545 0.8295 0.0679 0.043* 0.128 (4) H17A 1.1348 0.7653 0.0358 0.050* 0.872 (4) H17B 1.0791 0.9162 0.0454 0.050* 0.872 (4) H17C 0.8100 1.0096 0.0010 0.060* 0.128 (4) H17D 0.5670 0.9648 0.0170 0.060* 0.128 (4) H18 0.7611 0.8128 0.2802 0.027*
H19 0.6770 1.0327 0.2349 0.033* H20 0.3423 1.0283 0.1621 0.037* H21A 0.2594 1.2008 0.2675 0.046* H21B 0.0385 1.1399 0.2659 0.046* H22 0.1515 1.1129 0.4168 0.041* H23 0.5629 1.0841 0.3886 0.036* H24 0.6485 0.8615 0.4325 0.032* H26 −0.0048 0.9330 0.3802 0.035* H27 0.1072 0.8855 0.2338 0.030*
Atomic displacement parameters (Å2)
supporting information
sup-4
Acta Cryst. (2006). E62, o966–o968
C17 0.0447 (11) 0.0410 (10) 0.0390 (10) −0.0128 (8) −0.0024 (8) 0.0074 (8) C18 0.0195 (6) 0.0235 (6) 0.0254 (6) −0.0041 (5) −0.0005 (5) −0.0018 (5) C19 0.0245 (6) 0.0226 (6) 0.0354 (7) −0.0063 (5) 0.0002 (5) 0.0003 (5) C20 0.0297 (7) 0.0228 (6) 0.0386 (8) −0.0021 (5) −0.0050 (6) 0.0026 (5) C21 0.0327 (8) 0.0228 (6) 0.0594 (10) −0.0006 (5) −0.0052 (7) −0.0028 (6) C22 0.0284 (7) 0.0253 (7) 0.0477 (9) −0.0028 (5) 0.0026 (6) −0.0107 (6) C23 0.0274 (7) 0.0254 (6) 0.0393 (8) −0.0075 (5) −0.0013 (6) −0.0076 (6) C24 0.0242 (6) 0.0288 (6) 0.0269 (7) −0.0071 (5) −0.0011 (5) −0.0046 (5) C25 0.0275 (7) 0.0281 (6) 0.0286 (7) −0.0076 (5) 0.0032 (5) −0.0086 (5) C26 0.0212 (6) 0.0276 (6) 0.0369 (8) −0.0038 (5) 0.0035 (5) −0.0072 (6) C27 0.0203 (6) 0.0235 (6) 0.0312 (7) −0.0014 (5) −0.0043 (5) −0.0010 (5) C28 0.0197 (6) 0.0212 (6) 0.0215 (6) −0.0035 (4) −0.0018 (4) −0.0003 (5)
Geometric parameters (Å, º)
supporting information
sup-5
Acta Cryst. (2006). E62, o966–o968
C25—C24 1.5185 (18) C23—H23 1.0000 C26—C22 1.5585 (19) C24—H24 1.0000 C26—C25 1.514 (2) C26—H26 1.0000 C27—C20 1.5504 (18) C27—H27 1.0000 C27—C26 1.5805 (19)
supporting information
sup-6
Acta Cryst. (2006). E62, o966–o968
C5—C4—C3 95.36 (10) C23—C24—H24 116.6 C5—C4—H4A 112.7 C18—C24—H24 116.6 C3—C4—H4A 112.7 C28—C18—C19 104.41 (10) C5—C4—H4B 112.7 C28—C18—C24 111.12 (10) C3—C4—H4B 112.7 C19—C18—C24 90.59 (10) H4A—C4—H4B 110.2 C28—C18—H18 115.9 C4—C3—C10 104.38 (10) C19—C18—H18 115.9 C4—C3—C2 103.61 (11) C24—C18—H18 115.9 C10—C3—C2 100.11 (10) C21—C20—C19 103.15 (12) C4—C3—H3 115.6 C21—C20—C27 104.19 (12) C10—C3—H3 115.6 C19—C20—C27 100.65 (10) C2—C3—H3 115.6 C21—C20—H20 115.6 C3—C2—C6 102.85 (10) C19—C20—H20 115.6 C3—C2—C1 107.98 (10) C27—C20—H20 115.6 C6—C2—C1 89.84 (9) C22—C21—C20 95.83 (11) C3—C2—H2 117.4 C22—C21—H21A 112.6 C6—C2—H2 117.4 C20—C21—H21A 112.6 C1—C2—H2 117.4 C22—C21—H21B 112.6 C5—C6—C2 103.02 (10) C20—C21—H21B 112.6 C5—C6—C7 108.22 (10) H21A—C21—H21B 110.1 C2—C6—C7 89.90 (9) C21—C22—C26 104.19 (12) C5—C6—H6 117.3 C21—C22—C23 102.93 (12) C2—C6—H6 117.3 C26—C22—C23 100.88 (10) C7—C6—H6 117.3 C21—C22—H22 115.6 C16—C15—C8 112.31 (11) C26—C22—H22 115.6 C16—C15—H15A 109.1 C23—C22—H22 115.6 C8—C15—H15A 109.1 C22—C23—C19 102.90 (11) C16—C15—H15B 109.1 C22—C23—C24 107.69 (11) C8—C15—H15B 109.1 C19—C23—C24 90.37 (10) H15A—C15—H15B 107.9 C22—C23—H23 117.3 C17—C16—C17B 106.4 (4) C19—C23—H23 117.3 C17—C16—C15 124.54 (15) C24—C23—H23 117.3 C17B—C16—C15 124.50 (18) C20—C19—C18 107.80 (10) C17—C16—H16A 117.7 C20—C19—C23 103.06 (11) C15—C16—H16A 117.7 C18—C19—C23 89.84 (10) C17B—C16—H16B 117.8 C20—C19—H19 117.4 C15—C16—H16B 117.7 C18—C19—H19 117.4 H16A—C16—H16B 118.5 C23—C19—H19 117.4 C16—C17—H17B 119.5 C28—O1—C8 121.13 (9) H16B—C17—H17B 115.0 C28—O2—C11 119.74 (9) C16—C17—H17A 120.5
supporting information
sup-7
Acta Cryst. (2006). E62, o966–o968
supporting information
sup-8
Acta Cryst. (2006). E62, o966–o968

![Pentacyclo[5 4 0 02,6 03,10 05,9]undecane 8,11 dione ethylene acetal](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)