Thermodynamic Properties of Polybrominated Dibenzo-
p
-dioxins
and Dibenzofurans Calculated by Density Functional Theory
Xian-Wei Li
*, Etsuro Shibata and Takashi Nakamura
Institute of Multidisciplinary Research for Advanced Materials (IMRAM), Tohoku University, Sendai 980-8577, Japan
Heat capacities and entropies for 76 polybrominated dibenzo-p-dioxins (PBDDs) and 136 polybrominated dibenzofurans (PBDFs) in the gas state have been computed using the density functional theory. Based on the output data of Gaussian, three methods were employed to calculate enthalpies and Gibbs energies of formation of PBDDs and PBDFs in the gaseous state at 298.15 K and 101.325 kPa. To assess the three methods, thermodynamic properties of 16 brominated arenes compounds were first calculated and compared with experimental values. Among the three methods used, method 2 has the smallest average absolute deviation from the experimental data. All values for the heat capacity, entropy, enthalpy and energy of formation of the 76 PBDDs increase, as the number of substituted bromines increases. For isomers of tetrabromodibenzo-p-dioxins, 1,3,6,8-TeBDD, 1,3,7,8-TeBDD, 1,3,7,9-TeBDD and the most toxic compound 2,3,7,8-TeBDD are more stable than the others, and easier to form during formation process. Comparing with PBDDs, the formation enthalpies and Gibbs energies of PBDF isomers are more variable. The formation enthalpies and Gibbs energies of isomers which have bromine substitutions in 1 and 9 positions are much higher than those of the others.
(Received January 28, 2003; Accepted March 17, 2003)
Keywords: polybrominated dibenzo-p-dioxins (PBDDs), polybrominated dibenzofurans (PBDFs), thermodynamics, Heat capacity, Entropy, Enthalpy of formation, Gibbs energy of formation, density functional theory (DFT)
1. Introduction
Polybrominated dibenzo-p-dioxins (PBDDs) and
polybro-minated dibenzofurans (PBDFs) are dioxin congeners, they are persistent environmental contaminations. As concluded by the World Health Organization (WHO), PBDDs and PBDFs are more or less similar to polychlorinated
dibenzo-p-dioxins (PCDDs) and polychlorinated dibenzofuran
(PCDFs) in their persistence and toxicity.1)
PBDDs/PBDFs can be formed in various processes, the following potential cases have been identified as the release of PBDDs/PBDFs into environment.
. Formation during disposal and recycling of plastics
such as parts of office machine casings, printed circuit boards, scrap of electronic devices and cables.
. Formation during energy recovery by incineration of
waste plastics and utilizing waste plastics as blast furnace fuel.
. Formation from the laboratory thermolysis of
bromine-containing flame retardants.
. Formation during production of plastic materials and
presence in consumer products containing flame retar-dants, such as resins and polymer products.
. Emissions from flame-retarded consumer products. For
example, PBDFs were released from television sets, computers or similar appliances.
. Presence in fire residues, smoke condensates and gases
after fires. Both of experimental fires and accidental fires.
. By-products of brominated organic chemicals
(includ-ing flame retardants).
. Formation from the photochemical degradation of
brominated organic chemicals.
. Presence in automotive exhaust.
. Formation during textile processing.
Brominated flame retardants (BFR) and their precursors appear to be a main source of PBDDs/PBDFs. BFR including tetra-bromobisphenol A (TBBPA), polybrominated diphenyl ethers (PBDEs), polybrominated biphenyls (PBBs), and hexabromocyclodecane (HBCD) have been widely used in plastics, textiles, electronic circuitry and other materials to prevent fires, and hold an important market share. For example about 49000 and 64000 ton of BFR were processed
in Japan and in USA in 1999, respectively.2,3) Recycling
activities on these consumer products containing BFR are increasing and becoming more and more important in recent years, due to the formation of PBDDs/PBDFs in case of thermal stress. PBDDs/PBDFs were present in these materi-als of several recycling stages.
There is much less information on PBDDs/PBDFs than on their chlorinated analogues, and there are very few experi-mental data on their physical and chemical properties. The analytical methods for separating and identifying the indivi-dual brominated congeners are much less advanced than those for their chlorinated analogues, and only few reference standards are available. Current analytical methods are able to quantify total brominated homologue groups and also to detect but not quantify the mixed brominated/chlorinated congeners. Because of the complexity of analytical proce-dures and lack of reference standards, it has been possible to characterize and determine only a small number of PBDDs/ PBDFs and PXDDs/PXDFs, and only a few of the com-pounds have CAS registry numbers.
In this study, the thermodynamic properties (heat capacity, entropy, enthalpy and Gibbs energy of formation) in the gaseous state were computed for all 76 PBDDs and 136 PBDFs using density functional theory (DFT) methods with
Gaussian 98 programs.4) The purpose of the study was to
obtain a consistent set of thermodynamic values for PBDDs/ PBDFs. The discrepancy between the calculated results and available experimental values for 16 compounds (brominated
*Corresponding author: E-mail address: [email protected]
arenes) is also discussed. The present thermodynamic data are, to our knowledge, the first set of calculated data reported
on PBDDs/PBDFs.30)
2. Computational Methods
The theoretical calculations using DFT methods were carried out with the Gaussian 98 programs. Becke’s three-parameter hybrid functional combined with the gradient-correlation functional of Lee, Yang and Parr (LYP), denoted B3LYP, was employed in the computations using DFT. The all-electron 6-31G(d) basis set was employed. Geometries were optimized using analytic gradient techniques, i.e. the Berny algorithm with redundant internal coordinates. The stationary points on the potential energy surface were characterized by calculations of vibrational frequencies, which were done analytically at DFT levels. Following the geometry optimization, frequencies were calculated using the same method at a stationary point. The zero-point vibrational energies (ZPE) calculated at the DFT level were scaled by 0.9804.5)
Throughout this paper, all calculations for PBDDs/PBDFs were carried out with B3LYP/6-31G(d) Opt Freq. ‘‘Opt Freq’’ means that the frequency is calculated after
optimiza-tion of molecular geometry.
At present, this computational model level is higher than those which have been applied to calculate the thermody-namic values of dioxin congeners serially.6–8)
The equations used for computing thermochemical data in Gaussian programs are derived from statistical thermody-namics. Two key ideas of statistical thermodynamics are the Boltzmann distribution and the partition function. The partition function is like a thermodynamic wavefunction, in the sense that it contains all thermodynamic information about the system, just as the quantum mechanical wavefunc-tion contains all dynamic informawavefunc-tion.
2.1 Entropy
The entropy and heat capacity can be directly obtained from the output of Gaussian programs. The equations used for calculating the absolute entropy of molecule are as the following.9,10)
S¼StransþSrotþSvib ð1Þ
where Strans, Srot and Svib are translational, rotational and vibrational entropy, respectively, which can be calculated by the following equations:
Strans¼R
3 2ln
2mkT h2
þlnkT
p þ 5 2
ð2Þ
Srot¼R ln 1=2
r
þ1
2ln
T3
ðh2=82I
xkÞðh2=82IykÞðh2=82IzkÞ
þ3
2
ð3Þ
Svib ¼R X 3N6
i¼1
hv=kT
expðhv=kTÞ 1lnfexpðhv=kTÞg
ð4Þ
whereRis the gas constant (8.31451 Jmol1K1),Nis the atom number in a molecule,mis the molecular mass,kis the Boltzmann constant (1:3806581023JK1),his the Planck’s constant (6:62607551034Js),Tis the temperature,pis the pressure,r is the symmetry number for rotation,Iis the moment of inertia, andvis the vibrational frequency.
2.2 Heat capacity of constant pressure
The heat capacity at constant pressure was calculated using the following relation.9,10)
Cp ¼CtransþCrotþCvib¼
5 2Rþ
3 2RþR
X 3N6
i¼1
expðhv=kTÞ hv=kT
expðhv=kTÞ 1
2
ð5Þ
where Ctrans,Crot and Cvib are contribution to heat capacity due to translation, rotational motion, and vibrational motion, respectively.
2.3 Enthalpy and Gibbs energy of formation
The following equations are employed to calculate the absolute internal energy (U), enthalpy (H) and Gibbs energy (G) of the molecule at 0 K and the specified temperature (T).9,10)
U0 K¼EelecþEzpe ð6Þ
UT¼U0 Kþ ðEtransþErotþEvibÞT
¼ ðEelecþEzpeÞ þ
3
2RTþRTþR
X 3N6
i¼1
ðhv=kÞ 1
2þ
1
expðhv=kTÞ 1
( )
ð7Þ
HT¼UTþRT ð8Þ
GT¼HTTS ð9Þ
In this study,Eelecis computed at the B3LYP level.Etrans,
Erot and Evib can be rapidly calculated using statistical thermodynamics. All the values ofEelec,Ezpe,UT,HTandGT
are given in Hartrees (atomic units, 1 Hartree =
2625.51 kJmol1) by the output of the program.
Based on these absolute energy values (U,G,H), enthalpy and Gibbs energy of formation can be calculated by different methods.
Method 1
The enthalpies of formation at 0 K were calculated by subtracting the calculated atomization energies (D0) from known enthalpies of formation of the isolated atoms. The
enthalpies of formation at 298.15 K were calculated by correction to the enthalpies of formation at 0 K. This method is the common theoretical method for calculating the enthalpy of formation used by many studies.8,11–13)
For the computation of enthalpies of formation, Curtisset
al.13) tested seven density functional methods: B3LYP,
BP86, B3P86, BPW91, B3PW91 and SVWN with 148 molecules. Of these seven DFT methods, the B3LYP method has the smallest average absolute deviation (13.0 kJmol1) from the experimental values.
The calculation procedure is as follows:
fHðM;0 KÞ ¼ X
xfHðX;0 KÞ X
D0ðMÞ
¼XxfHðX;0 KÞ
X
xUðX;0 KÞ UðM;0 KÞ
h i
ð10Þ
fHðM;298 KÞ ¼fHðM;0 KÞ þ ½HðM;298 KÞ HðM;0 KÞ
XxðH298 KH0 KÞX ð11Þ
fGðM;298 KÞ ¼fHðM;298 KÞ TS
¼fHðM;298 KÞ T SðM;298 KÞ X
xSðX;298 KÞ
h i
ð12Þ
where fH andfG are the standard-state enthalpy and
Gibbs energy of formation of the ideal gas, respectively.M
stands for the molecule of the compound, X identifies each
element which consists of M, and x is the stoichiometric
coefficient of the constituent. ðH298 KH0 KÞX is the
forma-tion enthalpy correcforma-tion from 0 K to 298 K for elements in reference state.
fHðXÞ,S(X, 298 K), andðH298 KH0 KÞX are tabulated
in Table1, cited from the NIST-JANAF Thermochemical
Tables.14)The absolute standard state entropy S(X, 298 K) used for elemental carbon, hydrogen, bromine, and oxygen (reference state) should be (5.740, 130.680/2, 152.206/2, and 205.147/2) Jmol1K1respectively, not the values cited in Ochterski’s paper11)(not in reference state).
The calculated thermochemistry values using B3LYP/6-31G(d) for C, H, Br, O, H2, Br2, dibenzo-p-dioxin (DD) and
2,3,7,8-tetrabromodibenzo-p-dioxin (TBDD) are listed in
[image:3.595.45.549.521.609.2]Table2, all values are in Hartrees.
Table 1 Enthalpies of formation for gaseous atoms and entropy and (H298KH0K) values for elements in their reference state from
experiments.a
Atoms State fH
(0 K)
fH(298 K)
State S
(298 K) H
298KH0K
kJmol1 kJmol1 Jmol1K1 kJmol1
C Gas 711:190:46 716:670:46 Reference state 5:740:21 1.051 H Gas 216:0350:006 217:9990:006 Reference state 65:3400:017 4.238 Br Gas 117:920:06 111:860:06 Reference state 76.103 12.255
O Gas 246:790:10 249:170:10 Reference state 102:5740:018 4.342 N Gas 470:820:10 472:680:10 Reference state 95:8050:010 4.335
[image:3.595.44.549.644.756.2]aChase, 199814)
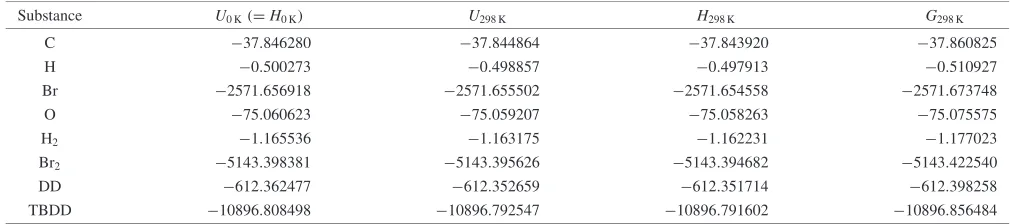
Table 2 Calculated thermochemistry values in gas phase at 101.325 kPa by B3LYP/6-31G(d). (Hartree)
Substance U0K(¼H0K) U298K H298K G298K
C 37:846280 37:844864 37:843920 37:860825
H 0:500273 0:498857 0:497913 0:510927
Br 2571:656918 2571:655502 2571:654558 2571:673748
O 75:060623 75:059207 75:058263 75:075575
H2 1:165536 1:163175 1:162231 1:177023
Br2 5143:398381 5143:395626 5143:394682 5143:422540
DD 612:362477 612:352659 612:351714 612:398258
TBDD 10896:808498 10896:792547 10896:791602 10896:856484
U0KandH0Kare the absolute internal energy and enthalpy of the molecule at 0 K.
U298K, H298K and G298K are the absolute internal energy, enthalpy and Gibbs energy of the molecule at 298.15 K, respectively.
Method 1 is illustrated with the example calculations for TBDD (C12H4Br4O2) as follows:
D0ðTBDDÞ ¼ ½12UðC;0 KÞ þ4UðH;0 KÞ þ4UðBr;0 KÞ þ2UðO;0 KÞ UðTBDD;0 KÞ
¼ ½12 ð37:846280Þ þ4 ð0:500273Þ þ4 ð2571:656918Þ þ2 ð75:060623Þ
ð10896:808498Þ ¼3.903128 Hartree¼10247:7 kJmol1
fHðTBDD;0 KÞ ¼ ½12fHðC;0 KÞ þ4fHðH;0 KÞ þ4fHðBr;0 KÞ þ2fHðO;0 KÞ D0ðTBDDÞ
¼ ð12711:19þ4216:035þ4117:92þ2246:790Þ 10247:7¼115:9 kJmol1
fHðTBDD;298 KÞ ¼fHðTBDD;0 KÞ þ ½HðTBDD;298 KÞ HðTBDD;0 KÞ ½12 ðH298 KH0 KÞC
þ4 ðH298 KH0 KÞHþ4 ðH298 KH0 KÞBrþ2 ðH298 KH0 KÞO
¼115:9þ ½ð10896:791602Þ ð10896:808498Þ 2625:51
ð121:051þ44:238þ412:255þ24:342Þ ¼73:0 kJmol1
fGðTBDD;298 KÞ ¼fHðTBDD;298 KÞ 298:15 ½SðTBDD;298 KÞ 12SðC;298 KÞ
4SðH;298 KÞ 4SðBr;298 KÞ 2SðO;298 KÞ
¼73:0298:15 ð571:3125:740465:340476:1032102:574Þ=1000
¼153:1 kJmol1
Method 2
Using B3LYP/6-31G(d), it was found that the enthalpy of formation results for benzene and DD calculated by Method 1 differ greatly from the experimental data. Therefore, a simple method, Method 2, was proposed here.
Because the absolute enthalpy (H) and Gibbs energy (G) values of the molecule can be obtained through theoretical calculation, it is easy to obtain the reaction enthalpy and Gibbs energy for any reaction using these energy values by eqs. (13) and (15). In another way, the reaction enthalpy and
Gibbs energy can be calculated by eqs. (14) and (16),
respectively.
rHð298 KÞ ¼ X
ðH298 KÞproducts X
ðH298 KÞreactants
ð13Þ
rHð298 KÞ ¼ X
ðfH298 K Þproducts X
ðfH298 K Þreactants
ð14Þ
rGð298 KÞ ¼ X
ðG298 KÞproducts X
ðG298 KÞreactants
ð15Þ
rGð298 KÞ ¼ X
ðfG298 KÞproducts X
ðfG298 KÞreactants
ð16Þ
Combining these equations and using the experimental data of enthalpy and Gibbs energy of formation for H2, Br2,
and DD,14,15) the unknown enthalpy and Gibbs energy of
formation values of TBDD can be calculated from the
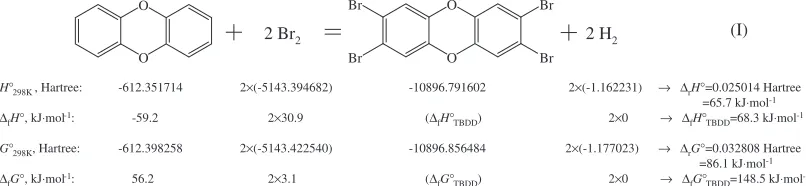
reaction (I) in Fig.1. Figure 1 shows the calculation
procedure of Method 2 (all reactants and products are in
gas state).
The of enthalpy and Gibbs energy of formation values of
TBDD calculated from reaction (I) are 68.3 kJmol1 and
148.5 kJmol1, respectively. Method 3(Benson’s method)
The third method for estimating the enthalpies of forma-tion is consistent with the group additivity technique developed by Benson.16)It is a traditional empirical method. Benson group values have been substantially refined during
the years,e.g.the CHETAH program17)by ASTM
Interna-tional predicts thermochemical properties using a modern Benson application. The available values of group contribu-tions to enthalpy of formation given by CHETAH 7.3 are listed in Table 3. For TBDD
fHTBDD ¼4ðCbHÞ þ4ðCbBrÞ þ4½Cb-(O)
þ2½O ðCbÞ2 þringþ2ortho
¼413:807þ444:769þ4 ð3:766Þ
þ2 ð78:659Þ þ8:368þ23:138
¼76:6 kJmol1
3. Results and Discussion
To assess the accuracy of the three methods used to predict the enthalpy and Gibbs energy of formation, the thermo-dynamic properties of 16 compounds (brominated arenes) were first calculated, and compared with available experi-mental data.
H°298K , Hartree: -612.351714 2×(-5143.394682) -10896.791602 2×(-1.162231) → ∆rH°=0.025014 Hartree =65.7 kJ⋅mol-1
∆fH°, kJ⋅mol-1: -59.2 2×30.9 (∆fH°TBDD) 2×0 → ∆fH°TBDD=68.3 kJ⋅mol-1
G°298K, Hartree: -612.398258 2×(-5143.422540) -10896.856484 2×(-1.177023) → ∆rG°=0.032808 Hartree =86.1 kJ⋅mol-1
∆fG°, kJ⋅mol-1: 56.2 2×3.1 (∆fG°TBDD) 2×0 → ∆fG°TBDD=148.5 kJ⋅mol-1 O
O
O
O
Br
Br Br
Br
2 Br2 2 H2 (I)
[image:4.595.95.498.673.766.2]3.1 Discrepancy analysis for the computation of ther-modynamics
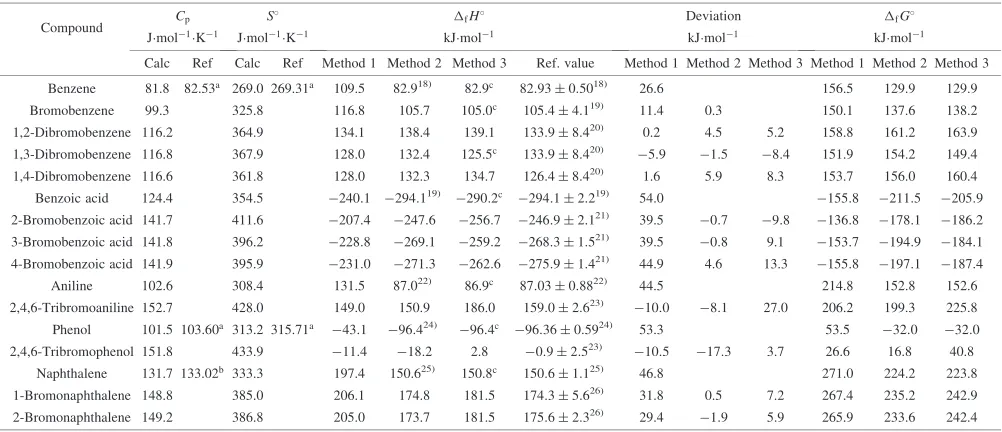
For brominated arenes, in fact, only minimal experimental thermodynamic data is available. Table4shows the calcula-tion results ofS,Cp,fHandfGfor benzene, bromoben-zenes, benzoic acid, bromobenzoic acids, naphthalene, and bromonaphthalenes in the standard-state ideal gas at 298.15 K and 101.325 kPa.
As shown, the calculated results of heat capacity and absolute entropy are in good agreement with experimental data, although little such data is available. On the properties of heat capacity and absolute entropy, the calculation results obtained by B3LYP/6-31G(d) seem to be accurate, since Gaussian employs the mature theoretical methods of statistics thermodynamics to compute these two thermodynamic properties, and this computational level is moderate.
In calculating the enthalpy of formation for those compounds, the results by Method 1 differ from the experimental values. The absolute deviations are from (10:5 to 54.0) kJmol1, and the average deviation is 28.1 kJmol1. The reason is the model chemistry (B3LYP/6-31G(d)) employed due to the tradeoff of accuracy and cost is not accurate enough for the absolute internal energy calculation.
The enthalpy of formation values calculated using method 2 are in good agreement with the experimental data of reference compounds. The average absolute deviation from
experimental values by method 2 is 4.2 kJmol1, and the
largest absolute deviation is 17.3 kJmol1.
The predicting values using methods 3 are also in reasonable agreement with the experimental. The average absolute deviation from experimental values for Method 3 is
9.8 kJmol1, and the largest absolute deviation is
27.0 kJmol1. For the enthalpies of formation of
2,4,6-tribromoaniline and 2,4,6-tribromophenol, both Method 2 and 3 have large differences from the sole experimental values by Allot and Finch.23)Unfortunately, there is a lack of experimental data for bromobenzene; the reference values of
dibromobenzene listed in Table4 are also estimated values
by Olesiket al.20)
The results show that the traditional Benson’s method of group additivity (Method 3) is still one of the most accurate methods for estimating formation enthalpy, and calculation process are very simple and fast.
However, Benson’s method can only give a rough correction for cis-trans isomerization empirically. In esti-mating the enthalpies of isomers, Method 2 is superior to Benson’s method, although Method 1 and 2 is far more computationally expensive.
Compared with the selected experimental data, Method 2 has the smallest absolute deviation among the three methods under the condition of B3LYP/6-31G(d). As indicated by Foresman and Frisch,5)model chemistries that are known to be quite reliable for optimizing geometries can be quite poor at predicting absolute thermochemical properties (such as
absolute internal energyU, enthalpyHand Gibbs energyG
[image:5.595.47.545.85.129.2]of the molecule), but such methods could be quite accurate at
Table 4 Comparison between calculated thermodynamic parameters and reference data in gas phase at 298.15 K and 101.325 kPa.
Compound Cp S
fH Deviation fG
Jmol1K1 Jmol1K1 kJmol1 kJmol1 kJmol1
Calc Ref Calc Ref Method 1 Method 2 Method 3 Ref. value Method 1 Method 2 Method 3 Method 1 Method 2 Method 3 Benzene 81.8 82.53a 269.0 269.31a 109.5 82.918)
82.9c 82:930:5018)
26.6 156.5 129.9 129.9 Bromobenzene 99.3 325.8 116.8 105.7 105.0c 105:44:119) 11.4 0.3 150.1 137.6 138.2
1,2-Dibromobenzene 116.2 364.9 134.1 138.4 139.1 133:98:420) 0.2 4.5 5.2 158.8 161.2 163.9
1,3-Dibromobenzene 116.8 367.9 128.0 132.4 125.5c 133:98:420) 5:9 1:5 8:4 151.9 154.2 149.4
1,4-Dibromobenzene 116.6 361.8 128.0 132.3 134.7 126:48:420) 1.6 5.9 8.3 153.7 156.0 160.4
Benzoic acid 124.4 354.5 240:1 294:119) 290:2c 294:12:219)
54.0 155:8 211:5 205:9
2-Bromobenzoic acid 141.7 411.6 207:4 247:6 256:7 246:92:121) 39.5 0:7 9:8 136:8 178:1 186:2
3-Bromobenzoic acid 141.8 396.2 228:8 269:1 259:2 268:31:521) 39.5 0:8 9.1 153:7 194:9 184:1
4-Bromobenzoic acid 141.9 395.9 231:0 271:3 262:6 275:91:421) 44.9 4.6 13.3 155:8 197:1 187:4
Aniline 102.6 308.4 131.5 87.022) 86.9c 87:030:8822) 44.5 214.8 152.8 152.6
2,4,6-Tribromoaniline 152.7 428.0 149.0 150.9 186.0 159:02:623) 10:0 8:1 27.0 206.2 199.3 225.8
Phenol 101.5 103.60a 313.2 315.71a 43:1 96:424) 96:4c 96:360:5924) 53.3 53.5 32:0 32:0
2,4,6-Tribromophenol 151.8 433.9 11:4 18:2 2.8 0:92:523) 10:5 17:3 3.7 26.6 16.8 40.8
Naphthalene 131.7 133.02b 333.3 197.4 150.625) 150.8c 150:61:125) 46.8 271.0 224.2 223.8
[image:5.595.48.549.192.408.2]1-Bromonaphthalene 148.8 385.0 206.1 174.8 181.5 174:35:626) 31.8 0.5 7.2 267.4 235.2 242.9 2-Bromonaphthalene 149.2 386.8 205.0 173.7 181.5 175:62:326) 29.4 1:9 5.9 265.9 233.6 242.4 aBarin, 198927)bThermodynamics Research Center, 199728)cExperimental values, CHETAH 7.3, 200217)
Table 3 Valuesaof group additivity contributions to enthalpy of formation of PBDDs.
Group CbH CbBr Cb–(O) O–(Cb)2
Correction
ring ortho gauche cis
predicting other molecular properties, vibrational frequen-cies, and a variety of relative energy values: energy differences to similar molecules, reaction energies (such as
rHandrG) and so on. The main reason for Method 2 can offer more accurate results is that the systematic errors forU,
H andGin the method often cancel out across the systems
being compared. Another reason is due to its use of experimental values as benchmarks.
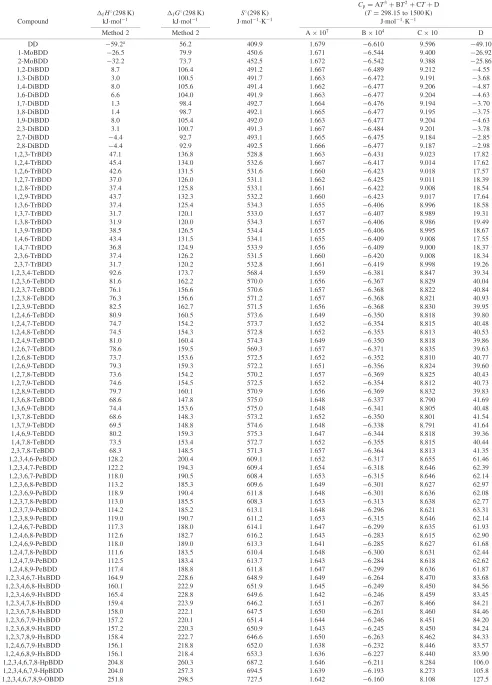
3.2 Calculation results of the thermodynamic properties of PBDDs/PBDFs
The calculation results of S, Cp, fH and fG for 76
PBDDs and 136 PBDFs in the gas phase at 298.15 K (forCp,
the temperature range is 298.15 to 1500 K) and 101.325 kPa
are listed in Tables 5 and 6 (for fH and fG, only the
values derived by Method 2 are listed in Tables5and6). As the substitute number of bromine increases, all the thermodynamic properties, namely the heat capacities, the absolute entropies, the formation enthalpies and the forma-tion Gibbs energies, of gaseous PBDDs and PBDFs increase. For predicting enthalpies and Gibbs energies of formation of PBDDs, the values obtained by Method 2 and Method 3 are in good agreement, except heptabromodibenzo-p-dioxins
and octabromodibenzo-p-dioxin. For low brominated
diben-zofuran, the enthalpies and Gibbs energies of formation values predicted by Method 2 and Method 3 are also in reasonable agreement.
The predicted enthalpy of formation values for low brominated (Br substitution number less than 4) dibenzo-p -dioxins and dibenzofuran by Method 1 are higher than those by Method 2, and the estimated values for high-brominated (Br substitution number greater than 4) dibenzo-p-dioxins and dibenzofuran by Method 1 are lower than those by Method 2. The values of Gibbs energy of formation thus have the same tendency.
Because of the lack of experimental data, the corrections for cis-trans isomerization used in Method 3 for PBDDs/ PBDFs are very rough, and the level sequence of energies of isomers is different from that predicted by Method 1 and Method 2, especially for PBDFs. In fact, Benzon’s method cannot distinguish energy differences among isomers.
Among 22 isomers of tetrabromodibenzo-p-dioxins, the
Gibbs energies of 1,3,6,8-tetrabromodibenzo-p-dioxin
(1,3,6,8-TeBDD), 1,3,7,8-tetrabromodibenzo-p-dioxin
(1,3,7,8-TeBDD), 1,3,7,9-tetrabromodibenzo-p-dioxin
(1,3,7,9-TeBDD), and 2,3,7,8-tetrabromodibenzo-p-dioxin
(TBDD, the most toxic compound in PBDDs) are lower than those of the other 18 isomers. It means that these 4 isomers are more stable, and easier to form during formation process. In the same way, 2,7-DiBDD and 2,8-DiBDD are easier to form than the other 8 isomers of dibromodibenzo-p-dioxins. 1,3,7-TrBDD, 1,3,8-TrBDD and 2,3,7-TrBDD are easier to
form than the other 11 isomers of tribromodibenzo-p
-dioxins. For the isomers of pentabromodibenzo-p-dioxins,
1,2,4,6,8-PeDBB, 1,2,4,7,8-PeDBB and 1,2,4,7,9-PeDBB are easier to form than the others. For the isomers of
hexabro-modibenzo-p-dioxins, 1,2,4,6,7,9-HxBDD and
1,2,4,6,8,9-HxBDD are easier to form than the others.
Comparing with PBDDs, the formation enthalpies and Gibbs energies of PBDF isomers are more variable. The
formation enthalpies and Gibbs energies of isomers which have bromine substitutions in 1 and 9 positions are much higher than those of the others. The reason is that the bromine atoms in 1 and 9 positions are close. The distance between the two bromine atoms in 1 and 9 positions is about
3:31010m, it is in the same level with the distance
between adjacent bromine atoms (ortho) in the same benzene ring.
The Gibbs energies of 1,7-dibromodibenzofuran
(1,7-DiBDF), 1,3,7-tribromodibenzofuran (1,3,7-TrBDF),
1,3,6,8-tetrabromodibenzofuran (1,3,6,8-TeBDF), 1,3,4,6,8-pentabromodibenzofuran (1,3,4,6,8-PeBDF) and 1,3,4,6,7,8-hexabromodibenzofuran (1,3,4,6,7,8-HxBDF) are the lowest among their isomers, respectively.
4. Conclusion
(1) Under the computing level of B3LYP/6-31G(d), Meth-od 2 has the smallest average absolute deviation and
maximum absolute deviation (4.2 and 17.3) kJmol1
from the experimental values of formation enthalpy of the selected reference compounds.
(2) Benson’s method is still a good method for estimating thermodynamic properties, and calculation procedures are very simple and much faster than Method 1 and 2. But, this method can only give much rougher correc-tions forcis-transisomerization empirically. Method 2 is superior to Benson’s method in predicting the formation enthalpies of isomers.
(3) All the heat capacity, entropy, enthalpy and Gibbs energy of formation values for the 76 PBDDs and 136 PBDFs increase as the substitute number of bromine increases. The values of enthalpy and Gibbs energy of formation of PBDDs and PBDFs calculated by Method 2 are recommended.
(4) For isomers of tetrabromodibenzo-p-dioxins, 1,3,6,8-TeBDD, 1,3,7,8-1,3,6,8-TeBDD, 1,3,7,9-TeBDD and the most toxic compound 2,3,7,8-TeBDD are more stable than the others, and easier to form during formation process. (5) Comparing with PBDDs, the formation enthalpies and Gibbs energies of PBDF isomers are more variable. The formation enthalpies and Gibbs energies of isomers which have bromine substitutions in 1 and 9 positions are much higher than those of the others.
Acknowledgments
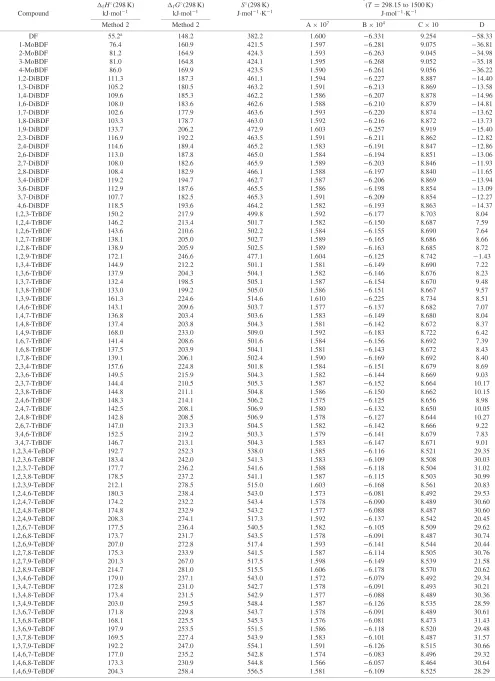
Table 5 Evaluated thermodynamic properties of gaseous PBDDs from 298.15 K to 1500 K at 101.325 kPa.
Cp¼AT3þBT2þCTþD fH(298 K) fG(298 K) S(298 K) (T¼298:15to 1500 K)
Compound kJmol1 kJmol1 Jmol1K1 Jmol1K1
Method 2 Method 2 A107 B104 C10 D
DD 59:2a 56.2 409.9 1.679 6:610 9.596 49:10
1-MoBDD 26:5 79.9 450.6 1.671 6:544 9.400 26:92
2-MoBDD 32:2 73.7 452.5 1.672 6:542 9.388 25:86
1,2-DiBDD 8.7 106.4 491.2 1.667 6:489 9.212 4:55
1,3-DiBDD 3.0 100.5 491.7 1.663 6:472 9.191 3:68
1,4-DiBDD 8.0 105.6 491.4 1.662 6:477 9.206 4:87
1,6-DiBDD 6.6 104.0 491.9 1.663 6:477 9.204 4:63
1,7-DiBDD 1.3 98.4 492.7 1.664 6:476 9.194 3:70
1,8-DiBDD 1.4 98.7 492.1 1.665 6:477 9.195 3:75
1,9-DiBDD 8.0 105.4 492.0 1.663 6:477 9.204 4:63
2,3-DiBDD 3.1 100.7 491.3 1.667 6:484 9.201 3:78
2,7-DiBDD 4:4 92.7 493.1 1.665 6:475 9.184 2:85
2,8-DiBDD 4:4 92.9 492.5 1.666 6:477 9.187 2:98
1,2,3-TrBDD 47.1 136.8 528.8 1.663 6:431 9.023 17.82 1,2,4-TrBDD 45.4 134.0 532.6 1.667 6:417 9.014 17.62 1,2,6-TrBDD 42.6 131.5 531.6 1.660 6:423 9.018 17.57 1,2,7-TrBDD 37.0 126.0 531.1 1.662 6:425 9.011 18.39 1,2,8-TrBDD 37.4 125.8 533.1 1.661 6:422 9.008 18.54 1,2,9-TrBDD 43.7 132.3 532.2 1.660 6:423 9.017 17.64 1,3,6-TrBDD 37.4 125.4 534.3 1.655 6:406 8.996 18.58 1,3,7-TrBDD 31.7 120.1 533.0 1.657 6:407 8.989 19.31 1,3,8-TrBDD 31.9 120.0 534.3 1.657 6:406 8.986 19.49 1,3,9-TrBDD 38.5 126.5 534.4 1.655 6:406 8.995 18.67 1,4,6-TrBDD 43.4 131.5 534.1 1.655 6:409 9.008 17.55 1,4,7-TrBDD 36.8 124.9 533.9 1.656 6:409 9.000 18.37 2,3,6-TrBDD 37.4 126.2 531.5 1.660 6:420 9.008 18.34 2,3,7-TrBDD 31.7 120.2 532.8 1.661 6:419 8.998 19.26 1,2,3,4-TeBDD 92.6 173.7 568.4 1.659 6:381 8.847 39.34 1,2,3,6-TeBDD 81.6 162.2 570.0 1.656 6:367 8.829 40.04 1,2,3,7-TeBDD 76.1 156.6 570.6 1.657 6:368 8.822 40.84 1,2,3,8-TeBDD 76.3 156.6 571.2 1.657 6:368 8.821 40.93 1,2,3,9-TeBDD 82.5 162.7 571.5 1.656 6:368 8.830 39.95 1,2,4,6-TeBDD 80.9 160.5 573.6 1.649 6:350 8.818 39.80 1,2,4,7-TeBDD 74.7 154.2 573.7 1.652 6:354 8.815 40.48 1,2,4,8-TeBDD 74.5 154.3 572.8 1.652 6:353 8.813 40.53 1,2,4,9-TeBDD 81.0 160.4 574.3 1.649 6:350 8.818 39.86 1,2,6,7-TeBDD 78.6 159.5 569.3 1.657 6:371 8.835 39.63 1,2,6,8-TeBDD 73.7 153.6 572.5 1.652 6:352 8.810 40.77 1,2,6,9-TeBDD 79.3 159.3 572.2 1.651 6:356 8.824 39.60 1,2,7,8-TeBDD 73.6 154.2 570.2 1.657 6:369 8.825 40.43 1,2,7,9-TeBDD 74.6 154.5 572.5 1.652 6:354 8.812 40.73 1,2,8,9-TeBDD 79.7 160.1 570.9 1.656 6:369 8.832 39.83 1,3,6,8-TeBDD 68.6 147.8 575.0 1.648 6:337 8.790 41.69 1,3,6,9-TeBDD 74.4 153.6 575.0 1.648 6:341 8.805 40.48 1,3,7,8-TeBDD 68.6 148.3 573.2 1.652 6:350 8.801 41.54 1,3,7,9-TeBDD 69.5 148.8 574.6 1.648 6:338 8.791 41.64 1,4,6,9-TeBDD 80.2 159.3 575.3 1.647 6:344 8.818 39.36 1,4,7,8-TeBDD 73.5 153.4 572.7 1.652 6:355 8.815 40.44 2,3,7,8-TeBDD 68.3 148.5 571.3 1.657 6:364 8.813 41.35 1,2,3,4,6-PeBDD 128.2 200.4 609.1 1.652 6:317 8.655 61.46 1,2,3,4,7-PeBDD 122.2 194.3 609.4 1.654 6:318 8.646 62.39 1,2,3,6,7-PeBDD 118.0 190.5 608.4 1.653 6:315 8.646 62.14 1,2,3,6,8-PeBDD 113.2 185.3 609.6 1.649 6:301 8.627 62.97 1,2,3,6,9-PeBDD 118.9 190.4 611.8 1.648 6:301 8.636 62.08 1,2,3,7,8-PeBDD 113.0 185.5 608.3 1.653 6:313 8.638 62.77 1,2,3,7,9-PeBDD 114.2 185.2 613.1 1.648 6:296 8.621 63.31 1,2,3,8,9-PeBDD 119.0 190.7 611.2 1.653 6:315 8.646 62.14 1,2,4,6,7-PeBDD 117.3 188.0 614.1 1.647 6:299 8.635 61.93 1,2,4,6,8-PeBDD 112.6 182.7 616.2 1.643 6:283 8.615 62.90 1,2,4,6,9-PeBDD 118.0 189.0 613.3 1.641 6:285 8.627 61.68 1,2,4,7,8-PeBDD 111.6 183.5 610.4 1.648 6:300 8.631 62.44 1,2,4,7,9-PeBDD 112.5 183.4 613.7 1.643 6:284 8.618 62.62 1,2,4,8,9-PeBDD 117.4 188.8 611.8 1.647 6:299 8.636 61.87 1,2,3,4,6,7-HxBDD 164.9 228.6 648.9 1.649 6:264 8.470 83.68 1,2,3,4,6,8-HxBDD 160.1 222.9 651.9 1.645 6:249 8.450 84.56 1,2,3,4,6,9-HxBDD 165.4 228.8 649.6 1.642 6:246 8.459 83.45 1,2,3,4,7,8-HxBDD 159.4 223.9 646.2 1.651 6:267 8.466 84.21 1,2,3,6,7,8-HxBDD 158.0 222.1 647.5 1.650 6:261 8.460 84.46 1,2,3,6,7,9-HxBDD 157.2 220.1 651.4 1.644 6:246 8.451 84.20 1,2,3,6,8,9-HxBDD 157.2 220.3 650.9 1.643 6:245 8.450 84.24 1,2,3,7,8,9-HxBDD 158.4 222.7 646.6 1.650 6:263 8.462 84.33 1,2,4,6,7,9-HxBDD 156.1 218.8 652.0 1.638 6:232 8.446 83.57 1,2,4,6,8,9-HxBDD 156.1 218.4 653.3 1.636 6:227 8.440 83.90 1,2,3,4,6,7,8-HpBDD 204.8 260.3 687.2 1.646 6:211 8.284 106.0 1,2,3,4,6,7,9-HpBDD 204.0 257.3 694.5 1.639 6:193 8.273 105.8 1,2,3,4,6,7,8,9-OBDD 251.8 298.5 727.5 1.642 6:160 8.108 127.5
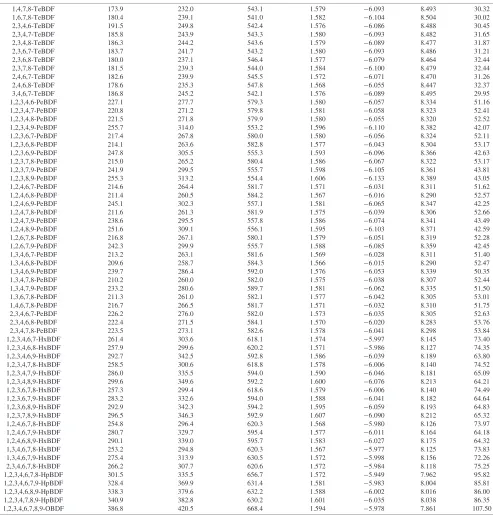
Table 6 Evaluated thermodynamic properties of gaseous PBDFs from 298.15 K to 1500 K at 101.325 kPa.
Cp¼AT3þBT2þCTþD fH(298 K) fG(298 K) S(298 K) (T¼298:15to 1500 K)
Compound kJmol1 kJmol1 Jmol1K1 Jmol1K1
Method 2 Method 2 A107 B104 C10 D
DF 55.2a 148.2 382.2 1.600 6:331 9.254 58:33
1-MoBDF 76.4 160.9 421.5 1.597 6:281 9.075 36:81
2-MoBDF 81.2 164.9 424.3 1.593 6:263 9.045 34:98
3-MoBDF 81.0 164.8 424.1 1.595 6:268 9.052 35:18
4-MoBDF 86.0 169.9 423.5 1.590 6:261 9.056 36:22
1,2-DiBDF 111.3 187.3 461.1 1.594 6:227 8.887 14:40
1,3-DiBDF 105.2 180.5 463.2 1.591 6:213 8.869 13:58
1,4-DiBDF 109.6 185.3 462.2 1.586 6:207 8.878 14:96
1,6-DiBDF 108.0 183.6 462.6 1.588 6:210 8.879 14:81
1,7-DiBDF 102.6 177.9 463.6 1.593 6:220 8.874 13:62
1,8-DiBDF 103.3 178.7 463.0 1.592 6:216 8.872 13:73
1,9-DiBDF 133.7 206.2 472.9 1.603 6:257 8.919 15:40
2,3-DiBDF 116.9 192.2 463.5 1.591 6:211 8.862 12:82
2,4-DiBDF 114.6 189.4 465.2 1.583 6:191 8.847 12:86
2,6-DiBDF 113.0 187.8 465.0 1.584 6:194 8.851 13:06
2,7-DiBDF 108.0 182.6 465.9 1.589 6:203 8.846 11:93
2,8-DiBDF 108.4 182.9 466.1 1.588 6:197 8.840 11:65
3,4-DiBDF 119.2 194.7 462.7 1.587 6:206 8.869 13:94
3,6-DiBDF 112.9 187.6 465.5 1.586 6:198 8.854 13:09
3,7-DiBDF 107.7 182.5 465.3 1.591 6:209 8.854 12:27
4,6-DiBDF 118.5 193.6 464.2 1.582 6:193 8.863 14:37
1,2,3-TrBDF 150.2 217.9 499.8 1.592 6:177 8.703 8.04 1,2,4-TrBDF 146.2 213.4 501.7 1.582 6:150 8.687 7.59 1,2,6-TrBDF 143.6 210.6 502.2 1.584 6:155 8.690 7.64 1,2,7-TrBDF 138.1 205.0 502.7 1.589 6:165 8.686 8.66 1,2,8-TrBDF 138.9 205.9 502.5 1.589 6:163 8.685 8.72 1,2,9-TrBDF 172.1 246.6 477.1 1.604 6:125 8.742 1:43
REFERENCES
1) WHO:Polybrominated Dibenzo-p-dioxins and Dibenzofurans,
Envir-onmental Health Criteria205, World Health Organization (Geneva,
1998) p. 223.
2) S. Sakai: http://www.nies.go.jp/sympo/2001/lecture/01-sakai/in-dex.htm
3) H. Wichmann, F. T. Dettmer and M. Bahadir: Chemosphere47(2002) 349–355.
4) . Frisch and M. J. Frisch:Gaussian 98 User’s Reference; second ed. (Gaussian, Inc., Pittsburgh, 1999) pp. 71–76.
5) J. B. Foresman and. Frisch:Exploring Chemistry with Electronic
Structure Methods, second ed. (Gaussian, Inc., Pittsburgh, 1996)
pp. 64–146.
6) C. J. Koester and R. A. Hites: Chemosphere17(1988) 2355–2362.
7) C. L. Huang, B. K. Harrison, J. Madura and J. Dolfing: Environ. Toxicol. Chem.15(1996) 824–836.
8) N. Saito and A. Fuwa: Chemosphere40(2000) 131–145.
9) D. A. McQuarrie and J. D. Simon: Molecular Thermodynamics, (Sausalito, Calif., University Science Books, 1999) pp. 1–656. 10) W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople:AB initio
Molecular Orbital Theory, (New York: Wiley. 1986) pp. 226–261.
11) J. W. Ochterski:Thermochemistry in Gaussian. http://www.gaussian.com/ thermo/thermo.pdf, 2000.
12) L. A. Curtiss, K. Raghavachari, P. W. Deutsch and J. A. Pople: J. Chem. Phys.95(1991) 2433–2444.
13) L. A. Curtiss, K. Raghavachari, P. C. Redfern and J. A. Pople: J. Chem. Phys.106(1997) 1063–1079.
[image:9.595.50.543.80.596.2]14) M. W. Chase:NIST-JANAF Thermochemical Tables, fourth edition, (J. Phys. Chem. Ref. Data 1998, Monograph No. 9, New York) pp. 470–1745.
Table 6 (Continued)
1,4,7,8-TeBDF 173.9 232.0 543.1 1.579 6:093 8.493 30.32 1,6,7,8-TeBDF 180.4 239.1 541.0 1.582 6:104 8.504 30.02 2,3,4,6-TeBDF 191.5 249.8 542.4 1.576 6:086 8.488 30.45 2,3,4,7-TeBDF 185.8 243.9 543.3 1.580 6:093 8.482 31.65 2,3,4,8-TeBDF 186.3 244.2 543.6 1.579 6:089 8.477 31.87 2,3,6,7-TeBDF 183.7 241.7 543.2 1.580 6:093 8.486 31.21 2,3,6,8-TeBDF 180.0 237.1 546.4 1.577 6:079 8.464 32.44 2,3,7,8-TeBDF 181.5 239.3 544.0 1.584 6:100 8.479 32.44 2,4,6,7-TeBDF 182.6 239.9 545.5 1.572 6:071 8.470 31.26 2,4,6,8-TeBDF 178.6 235.3 547.8 1.568 6:055 8.447 32.37 3,4,6,7-TeBDF 186.8 245.2 542.1 1.576 6:089 8.495 29.95 1,2,3,4,6-PeBDF 227.1 277.7 579.3 1.580 6:057 8.334 51.16 1,2,3,4,7-PeBDF 220.8 271.2 579.8 1.581 6:058 8.323 52.41 1,2,3,4,8-PeBDF 221.5 271.8 579.9 1.580 6:055 8.320 52.52 1,2,3,4,9-PeBDF 255.7 314.0 553.2 1.596 6:110 8.382 42.07 1,2,3,6,7-PeBDF 217.4 267.8 580.0 1.580 6:056 8.324 52.11 1,2,3,6,8-PeBDF 214.1 263.6 582.8 1.577 6:043 8.304 53.17 1,2,3,6,9-PeBDF 247.8 305.5 555.3 1.593 6:096 8.366 42.63 1,2,3,7,8-PeBDF 215.0 265.2 580.4 1.586 6:067 8.322 53.17 1,2,3,7,9-PeBDF 241.9 299.5 555.7 1.598 6:105 8.361 43.81 1,2,3,8,9-PeBDF 255.3 313.2 554.4 1.606 6:133 8.389 43.05 1,2,4,6,7-PeBDF 214.6 264.4 581.7 1.571 6:031 8.311 51.62 1,2,4,6,8-PeBDF 211.4 260.5 584.2 1.567 6:016 8.290 52.57 1,2,4,6,9-PeBDF 245.1 302.3 557.1 1.581 6:065 8.347 42.25 1,2,4,7,8-PeBDF 211.6 261.3 581.9 1.575 6:039 8.306 52.66 1,2,4,7,9-PeBDF 238.6 295.5 557.8 1.586 6:074 8.341 43.49 1,2,4,8,9-PeBDF 251.6 309.1 556.1 1.595 6:103 8.371 42.59 1,2,6,7,8-PeBDF 216.8 267.1 580.1 1.579 6:051 8.319 52.28 1,2,6,7,9-PeBDF 242.3 299.9 555.7 1.588 6:085 8.359 42.45 1,3,4,6,7-PeBDF 213.2 263.1 581.6 1.569 6:028 8.311 51.40 1,3,4,6,8-PeBDF 209.6 258.7 584.3 1.566 6:015 8.290 52.47 1,3,4,6,9-PeBDF 239.7 286.4 592.0 1.576 6:053 8.339 50.35 1,3,4,7,8-PeBDF 210.2 260.0 582.0 1.575 6:038 8.307 52.44 1,3,4,7,9-PeBDF 233.2 280.6 589.7 1.581 6:062 8.335 51.50 1,3,6,7,8-PeBDF 211.3 261.0 582.1 1.577 6:042 8.305 53.01 1,4,6,7,8-PeBDF 216.7 266.5 581.7 1.571 6:032 8.310 51.75 2,3,4,6,7-PeBDF 226.2 276.0 582.0 1.573 6:035 8.305 52.63 2,3,4,6,8-PeBDF 222.4 271.5 584.1 1.570 6:020 8.283 53.76 2,3,4,7,8-PeBDF 223.5 273.1 582.6 1.578 6:041 8.298 53.84 1,2,3,4,6,7-HxBDF 261.4 303.6 618.1 1.574 5:997 8.145 73.40 1,2,3,4,6,8-HxBDF 257.9 299.6 620.2 1.571 5:986 8.127 74.35 1,2,3,4,6,9-HxBDF 292.7 342.5 592.8 1.586 6:039 8.189 63.80 1,2,3,4,7,8-HxBDF 258.5 300.6 618.8 1.578 6:006 8.140 74.52 1,2,3,4,7,9-HxBDF 286.0 335.5 594.0 1.590 6:046 8.181 65.09 1,2,3,4,8,9-HxBDF 299.6 349.6 592.2 1.600 6:076 8.213 64.21 1,2,3,6,7,8-HxBDF 257.3 299.4 618.6 1.579 6:006 8.140 74.49 1,2,3,6,7,9-HxBDF 283.2 332.6 594.0 1.588 6:041 8.182 64.64 1,2,3,6,8,9-HxBDF 292.9 342.3 594.2 1.595 6:059 8.193 64.83 1,2,3,7,8,9-HxBDF 296.5 346.3 592.9 1.607 6:090 8.212 65.32 1,2,4,6,7,8-HxBDF 254.8 296.4 620.3 1.568 5:980 8.126 73.97 1,2,4,6,7,9-HxBDF 280.7 329.7 595.4 1.577 6:011 8.164 64.18 1,2,4,6,8,9-HxBDF 290.1 339.0 595.7 1.583 6:027 8.175 64.32 1,3,4,6,7,8-HxBDF 253.2 294.8 620.3 1.567 5:977 8.125 73.83 1,3,4,6,7,9-HxBDF 275.4 313.9 630.5 1.572 5:998 8.156 72.26 2,3,4,6,7,8-HxBDF 266.2 307.7 620.6 1.572 5:984 8.118 75.25 1,2,3,4,6,7,8-HpBDF 301.5 335.5 656.7 1.572 5:949 7.962 95.82 1,2,3,4,6,7,9-HpBDF 328.4 369.9 631.4 1.581 5:983 8.004 85.81 1,2,3,4,6,8,9-HpBDF 338.3 379.6 632.2 1.588 6:002 8.016 86.00 1,2,3,4,7,8,9-HpBDF 340.9 382.8 630.2 1.601 6:035 8.038 86.35 1,2,3,4,6,7,8,9-OBDF 386.8 420.5 668.4 1.594 5:978 7.861 107.50
15) V. P. Kolesov, O. V. Dorofeeva, V. S. Iorish, T. S. Papina, V. A. Luckyanova and S. V. Melkhanova: Organohalogen Compounds36
(1998) 201–204.
16) S. W. Benson:Thermochemical Kinetics, second ed. (John Wiley & Sons, New York, 1976) pp. 24–32.
17) ASTM International: The ASTM Computer Program for Chemical Thermodynamic and Energy Release Evaluation-CHETAH 7.3 User’s
Guide, (January 2002) pp. 27–31.
18) E. J. Prosen, W. H. Johnson and F. D. Rossini: J. Res. NBS36(1946) 455–461.
19) J. B. Pedley, R. D. Naylor and S. P. Kirby:Thermochemical Data of
Organic Compounds, 2nd ed. (Chapman and Hill, London, 1986)
pp. 116–215.
20) S. Olesik, T. Baer and J. C. Morrow: J. Phys. Chem.90(1986) 3563– 3568.
21) R. Sabbah and A. Rojas Aguilar: Struct. Chem.7(1996) 383–390. 22) W. E. Hatton, D. L. Hildenbrand, G. C. Sinke and D. R. Stull: J. Chem.
Eng. Data7(1962) 229–231.
23) P. H. Allot and A. Finch: J. Chem. Thermodyn.19(1987) 771–779. 24) J. D. Cox: Pure Appl. Chem.2(1961) 125–128.
25) D. J. Coleman and G. Pilcher: Trans. Faraday Soc.62(1966) 821–827. 26) M. A. V. Ribeiro da Silva, M. L. C. C. H. Ferrao and A. J. M. Lopes: J.
Chem. Thermodyn.25(1993) 229–235.
27) I. Barin:Thermochemical Data of Pure Substances, (VCH, Verlags Gesellschaft, Weinheim, 1989) pp. 241–267.
28) Thermodynamics Research Center:Selected Values of Properties of
Chemical Compounds, (Texas A&M University, College Station:
Texas, 1997).
29) R. D. Chirico, B. E. Gammon, S. E. Knipmeyer, A. Nguyen, M. M. Strube, C. Tsonopoulos and W. V. Steele: J. Chem. Thermodyn.22
(1990) 1075–1096.