warwick.ac.uk/lib-publications
A Thesis Submitted for the Degree of PhD at the University of Warwick
Permanent WRAP URL:
http://wrap.warwick.ac.uk/108018/
Copyright and reuse:
This thesis is made available online and is protected by original copyright.
Please scroll down to view the document itself.
Please refer to the repository record for this item for information to help you to cite it.
Our policy information is available from the repository home page.
INVESTIGATIONS O N ENZYME-CATALYZED
PEPTIDE SYNTHESIS
By
Jean-Marc RICCA
Submitted for the degree o f Doctor of Philosophy
University o f Warwick
3
-CONTENTS
Pages
Abbreviations 5
Acknowledgement 7
List o f figures 8
Declaration 10
Publication 11
Summary 12
General introduction 13
1- introduction 13
2- use o f enzymes in peptide synthesis 16
2-1. introduction 16
2-2. chronology 17
2-3. thermodynamically controlled peptide synthesis 19
2-4. kinetically controlled peptide synthesis 24
2-5. major developments in enzyme-catalyzed 26
peptide b ond formation
Chapter I : applications of Chymotrypsin suspended 39
in organic solvents
1-1. Peptide synthesis catalyzed b y chymotrypsin 39
in organic solvents
1-1.1. introduction 39
1-1.2. materials and methods 41
1-1.3. results and discussion 45
1-2. Hydrolytic reactions catalyzed b y chymotrypsin 83
suspended in organic solvents with low water content
1-2.2. materials and methods 85
I- 2.3. results and discussion 87
Chapter II : kinetics o f Chymotrypsin controlled syntheses 114
of peptide bonds in organic solvents
II-l. Introduction 114
II-2. Methods and experimental procedures 120
II-3. Results and discussion 122
II- 3.1. effect of the water content 122
II-3.2. kinetics o f chymotrypsin-catalyzed synthesis 130
o f N-acetyl-L-tyrosyl-L-phenylalaninamide in
dichloromethane
II-4. Conclusions 142
Chapter III : Chymotrypsinogen and Chymotrypsins as 145
catalysts for peptide synthesis
Chapter I V : Enzymatic synthesis of leucine-enkephalinamide 160
Chapter V : Design o f competitive inhibitors o f proteases 208
V-l. Introduction 208
V-2. Hydrolysis of carbon-carbon bonds b y Chymotrypsin 221
V-3. Synthesis o f 3-keto esters derived from amino acids 227
V-4. Diastereoselective reductions o f 3-keto esters 234
Experimental details 249
4
5 -ABBREVIATIONS z BOC Ac Bz Bzl Bu Ph Et Me NMe Mca FMOC OTMB ATOIi ATEE THF CDI MTPA PEG DMF DMSO EEDQ TFA CT CDP-Y PPL NAD(P)H HLADH RNA NCYC TMS IgE benzyloxycarbonyl tert-butyloxycarbonyl acetyl benzoyl benzyl butyl phenyl ethyl methyl methylamide monochloroace ty1
9-fluorenylmethyloxycarbonyl trimethylbenzyl ester N-acetyl-L-tyrosine ethyl N-acetyl-L-tyrosinate tetrahydrofuran carbonyldiimidazole (-)-2-methoxy-2-trifluoromethylphenylacetyl polyethylene glycol
dime thyl formamide dimethylsulfoxi.de
N-ethyloxycarbony1-2-ethyloxy-1,2-dihydroquinoline trifluoroacetic acid
Chymotrypsin carboxypeptidase Y pig pancreatic lipase
dihydronicotinamideadenine dinucleotide(phosphate) horse liver alcohol dehydrogenase
ribonucleic acid
national collection of yeast cultures tetramethylsilane iimiunoglobulin E NMR ppm J s d t q b NOE IR M c M P BP GLC HPLC hrs TLC
nuclear magnetic resonance parts per million coupling constant singlet doublet triplet quartet broad
nuclear Overhauser enhancement infrared
specific rotation concentration (g/100ml) melting point boiling point
gas-liquid chromatography
high performance liquid chromatography hours
6
-MS mass spectrometry
FAB fast atom bombardement
CID collision induced decomposition
ee enantiomeric excess
de diastereoisomeric excess
kcat maximal velocity per unit of enzyme concentration
K m substrate concentration at half-maximal velocity
Ki inhibition constant
Vsyn initial velocity for peptide bond formation
Vhyd initial velocity for hydrolysis
Notations o f the standard amino acids :
alanine Ala tyrosine Tyr
aspartic acid Asp cysteine Cys
leucine Leu tryptophan Trp
glycine Gly histidine His
lysine Lys ornithine O m
serine Ser valine Val
arginine Arg threonine Thr
proline Pro methionine Met
7
-I would like to thank Professor D.H.G. Crout for the guidance and
encouragement which h e has provided throughout the course o f this
work.
Further I want to thank Dr. K. Muller for the molecular modelling
studies.
I must also thank Dr. 0. Howard for his assistance w ith the NMR
studies and Dr. D. Despeyroux for the tandem mass spectrometry
studies.
Thanks are due to Dr. M. Donelly for the Karl-Fischer analysis of
the powdered enzymes and Dr. D . A Taylor for the electron
micrographs o f the chymotrypsin surfaces.
The financial support of Rhône-Poulenc Recherches is gratefully
LIST O F FIGURES
Chapter I
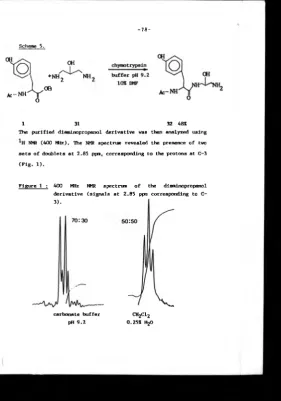
Figure 1. 400 M H z NMR spectrum o f the diaminopropanol
derivative.
Figure 2. The active site o f chymotrypsin according
to the model of Cohen.
Chapter II
Figure 1. Effect o f the water content (heterogeneous
catalysis).
Figure 2. Effect of the w a ter content (biphasic
conditions).
Figure 3. Effect of the water content for
D-nucleophiles (heterogeneous catalysis).
Figure 4. Effect of the water content for
D-nucleophiles (heterogeneous catalysis).
Figure 5. Initial velocities pattern as a function of
the concentration o f the starting ester.
Figure 6. Double-reciprocal Lineweaver-Burk plot
showing the initial velocity pattern with
ATEE a s the varied-concentration substrate.
Figure 7. composition o f the reaction mixture with
various concentrations o f the nucleophile
(0-160mM) and fixed concentrations o f ATEE
(80mM) and water ( H O m M ) (reaction time 20
minutes).
Figure 8. Variation o f Ln[Vsyn/(Vm-Vsyn) ] against
U \ PheNH2 .
Figure 9. Initial-velocity ratio o f peptide bond
formation to hydrolysis as a function of
substrate concentrations.
Chapter III
Figure 1. Activation scheme for the Chymotrypsins.
Figure 2. A v i e w o f the complete polypeptide c h ain of
a -Chymotrypsin.
9
-Chapter IV
Chapter V
hour.
Figure 1. Full mass spectrum of synthetic leucine-
enkephalinamide.
Figure 2. CID mass spectrum of synthetic leucine-
enkephal inamide.
Figure 3. Full mass spectrum of the enzymatically-
synthesized leucine-enkephalinamide.
Figure 4. CID mass spectrum corresponding to the ion
at m / z 689.3.
Figure 5. CID mass spectrum corresponding to the ion
at m / z 688.
Figure 6. FAB mass spectrum of PPL-catalyzed
synthesis of leucine-enkephalinamide.
Figure 1. Double-reciprocal Lineweaver-Burk plot
1 0
-T h e work described in this thesis is the original w ork o f the
aut h o r except where acknowledgement has been made to results and
ideas previously published. It was carried out in the Department
o f Chemistry, University of Warwick between October 1987 and
September 1990 and has not been submitted previously for a degree
1 1
-PUBLJCATION
Part o f the research described in this thesis has appeared in the
scientific literature as follows :
Peptide synthesis catalyzed b y Chymotrypsin in organic solvents.
J.M. Ricca, D.H.G. Crout, J. Chem. Soc., Perkin Coranun., 2126
1 2
-S I M M K Y
Enzymes have been found to be catalytically active in organic
solvents. Chymotrypsin was used to synthesize a wide range of
peptides when suspended in organic solvents. This method overcame
such problems as secondary hydrolysis and poor solubility in
aqueous mixtures, and allowed processes to occur that were
impossible in water. Syntheses involving D-amino acid derivatives
w ere possible under these conditions. Molecular modelling studies
h ave been carried out and structure-reactivity relationships have
b e e n drawn by using hydrolytic reactions catalyzed by
chymotrypsin suspended in organic solvents with low water
c o n t e n t .
K inetic studies o f chymotrypsin suspended in organic solvents
h a v e shown that the enzyme does not have a classical Michaelis-
Men t e n behaviour, but shows cooperative effects with respect to
the binding of the substrates. The role o f the essential water
h a s b een investigated.
T h e use of the different chymotrysins in peptide synthesis has
b e e n investigated and 7 -chymotrypsin, inactive in water, has
b e e n found fully active when suspended in organic solvents.
T h e enzymatic synthesis o f leucine-enkephalinamide was carried
out and tandem mass spectrometry was used to determine the
composition of the reaction mixture. The last coupling step used
a n enzyme in an organic solvent.
Potential competitive inhibitors o f proteases have been designed
a n d synthesized. Several methods for the synthesis o f 3-keto
esters w ere investigated and diastereoselective reductions o f 3-
1 3
-G S I E R A L INTRODUCTION
1 - Introduction
Tremendous advances in peptide synthetic chemistry have been made
s i nce the d a y w hen Qnil Fischer defined the peptide bond as the
amide-like linkage between amino acids and gave the reasons why
h e chose the t erm "peptide"
Peptides and proteins exhibit the largest structural and
functional variations o f all classes of biological
macromolecules • They are o f prime importance in the regulation
a n d maintenance of all biological processes. The essential
structural features o f peptide and protein molecules are chains
o f amino acids linked to one another b y amide bonds. Important
aspects and considerations are the polymeric nature of peptides
a n d proteins a n d their wide range o f properties. The polymeric
character requires the use o f special approaches to synthesis.
T h e spectrum o f peptide properties - from very basic to very
acidic, from highly hydrophilic to totally hydrophobic, from
e a s i l y soluble to completely insoluble - invariably presents
surprises and obstacles during synthesis o f these molecules.
M o d e r n synthetic peptide chemistry really started in 1953 with
the chemical synthesis of the nonapeptide hormone oxytocin by
1 4
-The m o s t frequently used methods of peptide synthesis are those
of a chemical nature. The chemical formation o f a peptide bond
in principle, can b e reduced to four steps (Scheme 1).
S c h e m e 1 : General scheme of peptide synthesis. Z, amine
protecting group; Y, carboxyl protecting group; X,
activating substituent.
. a * * , , * « * ,
R 0
Z -N H -C H -i-O H
carboxyl coaponent
t W SUSP : u r t a s J s d * * »
j
R o
Z - N H - C H - i - X
3 n1 stag« : peptide bond RruXun
R
r>
NH-CH-<?-o’
NHjCH-i-Y
Z - N H - C H - i -
NH-CH
-1
-t -tb s-tage : depro-tec-tion
1 5
-Side chains (R) o f certain amino acids contain functionalities
w h i c h n e e d protection that is maintained throughout the synthesis
and therefore termed semi-permanent protection. In the second
stage, the carboxyl group of the N-protected carboxyl component
is activated. "Coupling reagents" ^ conveniently effect selective
activa t i o n of the carboxyl component in the presence o f the amine
component. Peptide b o n d formation constitutes the third stage. An
efficient condensation procedure should provide for rapid peptide
b ond formation, m inimal racemization, few side reactions, ready
work-up, and high yields. The routine laboratory scale peptide
s y n thesis by solution techniques w ith u p to 15 amino acid
residues, can be considered as a realistic goal. Segment
condensation has b e e n successfully applied to the synthesis of
m a n y peptide hormones with up to about 60 amino acid residues.
T his limit will p r obably be extended b y further refinements or
exi s t i n g methods a n d developments o f novel approaches such as
"the f o u r component segment condensation" ^ or "the amine capture
method"
Fo l l o w i n g B. Merrifield's ingenious innovation o f covalently
b i n d i n g the growing peptide chain to an insoluble polymeric
support solid-phase methodology has become increasingly
p o p u l a r during the last 20 years. In this strategy, excess
r eagents and by-products from the synthetic cycles can be removed
b y s i m p l e filtration a n d washing steps. A reappraisal of solid-
phase peptide synthesis in the early 1970s ^ led to the
1 6
-Merrifield were not necessarily optimal. Higher reaction yields
might b e obtained if the special nature of the chemical
environment within the polymer matrix was considered. These
considerations led to the development of a n e w variant o f solid-
phase pep t i d e synthesis, n a m e l y the FMOC-polyamide method 8 . The
most attractive improvement brought about b y this procedural
simplification is the time-saving and convenient mode of
operation and consequently, the prospect o f fully automated
synthesis ^ .
Nevertheless, the considerable shortcomings of these methods
still impose a n undiminished challenge u pon synthetic peptide
chemistry. These limitations arise mainly from the fact that the
individual steps o f the synthetic pathway are relatively
unspecific in nature. Consequently, the success o f many syntheses
is jeopardized b y the appearance of undesired by-products. 2****
2- Use o f enzymes in peptide synthesis,
2-1. Introduction
Enzymes h a v e been w i d e l y used as catalysts in organic
synthesis Recent experiments in several laboratories have
1 7
-offering several advantages over chemical methods for the
formation o f peptide bonds for synthesis a n d semisynthesis.
The c a pacity o f proteases to effect peptide bond synthesis
stereospecifically and without the need o f side chain protection
was recognized early in the study o f these enzymes as a natural
result o f their catalytic nature Although these early
successes occurred largely from their being driven by
precipitation o f synthesized products, a m ore controlled type of
protease-catalyzed peptide b o n d formation was observed in the
ability o f carboxypeptidases A and B, trypsin, and chymotrypsin
to re-form specific peptide bon d s in trypsin inhibitors *2 .
2-2. CHRONOLOGY
The concept o f peptide synthesis b y reversal o f mass action in
protease-catalyzed reactions dates back to 1898, when J.H. Van't
Hoff supposed that the protease "trypsin" could be endowed w ith
the inherent capacity to catalyze the synthesis o f proteins from
degradation products originally generated b y its own proteolytic
action 13.
The rationale behind this idea involved the applicability o f the
law of m a s s action to enzyme controlled reactions and their
reversibility arising from the presuned catalytic nature o f the
1 8
-During the first decades of the present century many biochemists
believed that a biochemical process that required free energy to
take place c o u l d be accomplished w i t h the greatest efficiency by
living organisms. As a consequence, it was generally assumed
that, for instance, the catabolic pathways o f biological
macromolecules w e r e inversely equal to the anabolic ones.
Moreover, this v iew implicitly suggested the possibility of
preparing pr o t e i n s by "hydrolysis in reverse" via protease
catalysis. T h i s idea of protein biosynthesis b y "reversible
enzymic hydrolysis" had long been considered to be supported by
the phenomenon o f the so-called "plastein reaction". As early as
1901, Savjalov described plastein formation correctly as the
outcome o f a "proteosynthetic" process, namely as the reverse of
the already k n o w n "proteolytic" action of proteases Due to
their complexity, however, the chemical nature o f the plasteins
could not b e e x a c t l y characterized b y the methods o f that time.
After simplifying the experimental conditions, Bergmann's group
was the first t o describe the enzymatic syntheses o f well-defined
dipeptides v i a papain 15 and a -chymotrypsin catalysis
However, the concept of protein biosynthesis b y reversal of
enzymatic proteolysis, which was accepted as plausible until the
beginning o f the 1940's was put in question b y thermodynamic data
on peptide bond hydrolysis. It could be shown that the synthesis
o f peptide b o n d s represents a strongly endergonic process under
1 9
-"peptide bonds cannot be synthesized to a n y significant extent
merely b y mass action reversal o f hydrolysis".
The demise o f the concept o f protease-controlled protein
biosynthesis finally coincided with the recognition o f the
genetic code a n d the decisive role played b y mRNAs and tRNAs
during the process o f in vivo protein synthesis.
Besides their primary role in peptide synthesis, proteases have
also been successfully applied to oligomerization
semisynthesis and protecting group chemistry
2-3. Thermodynamically controlled peptide synthesis.
The thermodynamically controlled formation o f peptide bonds
represents the direct reversal o f the catalytic cleavage of
peptides b y proteases ^ .
Since, however, concentrations are used i n the description below
instead o f activities, this is not a n exact thermodynamic
treatment. I n contrast to the hydrolysis, the synthesis o f a
peptide bond is a n endergonic process, i.e., proceeds w ith loss
of entropy a n d is energetically so unfavorable that the
equilibriun constant Ksyn for the coupling o f two unprotected
2 0
-are unreaetive,for the thermodynamic approach, two equilibria
have to be t a ken into account :
R-COO" + +H 3N-R' M g S R-COOH + H 2N-R R-CONH-R' + H 20
Preceding the "inversion equilibriun" Kinv between the uncharged
substrates and the product is an "ionization equilibrium" Kion.
Taking the concentration o f water into the equilibrium constant,
the total process is given b y :
K s y n = K i o n .Kinv= (R C O N H R ' )((RCOO“ )(+H 3HNR'J)“ 1
For any given p a i r o f substrates and known pH, Kion and Kinv are
fixed. The o n l y function o f the protease is to accelerate the
attainment o f the equilibrium for the formation o f the peptide.
Therefore, r e action conditions should be cho s e n to ensure a high
catalytic a c t i v i t y of the protease. The p H optimum o f the
synthesis lies a p art from pepsin catalyzed couplings, in the pH
range between t h e p K of the a-carboxyl g r o u p and that o f the
amino group o f t h e substrates, i.e., normally between p H 6 and 7.
There are two principal ways b y which one c a n further influence
thermodynamically controlled peptide formation :
- increasing Kion b y alteration o f the p K values o f the
2 1
-- Increasing the concentration of the peptide product via
manipulation b a s e d on the law o f mass action.
2-3.1. Increase i n Kion
For a given set o f reaction conditions, there are two w ays to
decrease the difference in pK values between amino and carboxyl
components in o r d e r to increase Kion. This leads in turn, to an
increase in Ksyn = Kion . Kinv
2-3,1.1. Use o f water-miscible organic solvents
Water-miscible organic solvents decrease the acidity of the at-
carboxy group o f the carboxy component, whereas they only
marginally influence the pK value of the a m ino group o f the
nucleophile. For example, the p K value of acetylglycine in water
is 3.60, while i n 80X (v/v) dimethylsulfoxide it is 6.93 ; the pK
values of GlyNHjg 8.20 and 8.10, respectively, remain almost
constant. A v a r i e t y of other cosolvents can b e u sed to change the
pK value by one t o two units, resulting in a significant increase
in Ksyn. This approach, however, is problematic in so far as the
catalytic a c t i v i t y of proteases decreases with increasing
concentration o f the cosolvent ; thus, the time required to reach
the inversion o f the equilibrium increases. O n l y polyalcohols,
which act as enzyme stabilizers, may b e used at high
2 2
-2-3.1.2. Use of bip h a s i c systems
In systems consisting o f an aqueous phase and a n o n miscible
phase (non-polar o r g a n i c solvent), pK values are influenced in
such a way that K i o n increases Since the enzyme is localized
in the aqueous phase, the activity can be influenced only by the
saturation concentration o f the organic solvent in water, and the
enzyme is therefore inhibited far less than b y solvents miscible
with water. This advantage, however, is counteracted by the
prolonged time r e quired to reach equilibrium. Solubility of the
substrates in the n onpolar organic phase limits the general use
o f biphasic systems f o r the enzymatic peptide synthesis.
2-3.2. Influence o n product formation based on the law of Hass
Action
The first positive experimental results for the use o f a reversal
o f protease-catalyzed peptide hydrolysis were bas e d on the
limited solubility o f the products, which thus are removed from
the inversion e quilibrium (see p age 20)and accumulate ^ . The
product can also b e removed from the equilibrium b y extraction or
specific complexation.
2-3.2.1. Formation o f insoluble products
If sufficiently h i g h concentrations o f substrates a r e employed,
2 3
-peptide concentration that lies above the maximal saturation
concentration, precipitation occurs and thus the product o f the
synthesis accumulates. The apparent equilibrium constant, for a
reaction in which some o f the product precipitates d u e to its
limited solubility in the system, is greater the high e r the
starting concentration o f substrates and the lower the solubility
o f the product in this system. Using one component in excess may
result in almost quantitative reaction o f the other substrate.
This method is very popular in practice since the condition of
low solubility o f the product compared to that of the substrates
frequently holds.
2-3.2.2. Extraction o f products
In biphasic systems, the product is removed from the equilibriim
if, owing to a favorable position o f the equilibrium, it is
extracted and thus accumulates in a nonpolar phase that is not
miscible with water. I n most cases, however, the product is only
marginally soluble in the organic phase ; it precipitates and is
thus removed from the equilibrium.
2-3.2.3. Specific complexation of the product
If compounds are available that can form specific complexes with
the product, the latter can be removed f r o m the equilibrium by
2 4
-2-4, Kinetlcally controlled syntheses.
Investigation of the catalytic mechanism o f serine and cysteine
proteases using Chymotrypsin and papain a s examples revealed
that, in the presence o f nucleophiles, acyl enzymes intermediates
RCO-E are deacylated competitively b y water and the nucleophile
If the nucleophile is a n aminoacid or peptide derivative, then a
new peptide is formed during the aminolytic deacylation
(Scheme 2).
Scheme 2,
r
-
o o
-
n h
-
r
'*
h
-
e
l 4
t NH
2
-
r'
l2
1
R-COOX-H-E --- R -C O -E
2 5
-Especially suitable carboxyl components for this type of
enzymatically catalyzed peptide synthesis are acylamino acid
alkyl esters, if they match the substrate specificity of the
particular protease, since in general they fulfill the condition
k 2 3>k3+k^ . Furthermore, k 2 o f the ester substrate is
substantially greater than k 2 of the peptide formed, which
results in a maximum for product formation before the slower
hydrolysis o f the product starts to become important.
The area in which kinetically controlled synthesis c a n be used in
practice is limited to serine and cysteine proteases which prefer
acylamino acid esters as the carboxy component. The reactions are
characterized by short reaction times and low enzyme
requirements.
2-4.1. Influence of the reaction medium
Since only the non-protonated form o f the nucleophile reacts in
the amino lytic deacylation o f the acyl-enzyme, it is necessary to
take the effective concentration o f nucleophile instead of the
total concentration into account. This can be calculated from the
p K value o f the nucleophile and the pH o f the meditm. Since the
pK values of u-amino groups o f amino acid and peptide
derivatives lie around 8 it is advisable to carry o u t kinetically
2 6
-2-5. Ma.jor developments In enzyme-catalyzed peptide bond
formation.
2-5.1. Enzymatic reactions in aqueous-organic media
Recently, the area o f enzymatic reactions in the presence of
organic solvents, either miscible o r inmiscible with water, has
been rapidly growing and drawing m u c h attention. These reactions
are of great interest w ith respect to both basic studies o n the
medium effects o n enzyme catalysis and the application of
enzymatic reactions to organic synthesis. For example, hydrolytic
enzymes such as a -Chymotrypsin, h ave been employed as catalysts
for various ester syntheses i n aqueous-organic two-phase
systems 2®.It has been considered that in this system the
decreased amount o f water and the low solubility of the products
in the aqueous phase shift the reaction equilibriun towards ester
synthesis. However, w hen a hydrophobic (water-immiscible) organic
solvent is used, the m a i n drawback o f the method is that if one
of the reactant RCOOH such as ami n o acids is sparingly soluble in
the organic phase, it would be concentrated in the aqueous phase.
This often causes a substrate inhibition of the enzyme, leading
to low yields of the esters 2 ^»2®. The synthetic reactions by
hydrolytic enzymes i n water-hydrophilic (water-miscible) organic
solvents have also b een reported 29,30_ gy using limited amounts
of water in solvents, the equilibriun of the reaction can be
2 7
-the enzymes is often impaired at high concentrations o f organic
solvents, and the yields o f products have been rather limited.
For example, it has b een reported that N-acetyl-L-tyrosine ethyl
ester was obtained in 25-30% yields b y chymotrypsin-catalysed
reactions o f N-acetyl-L-tyrosine w ith ethanol in 50-60% water,
but that chymotrypsin was inactivated at lower concentrations of
water 3 1 . However, the dipeptide derivatives Z-L-Tyr-L-LeuNH2
and Mca-L-Tyr-L-LeuNH2 were synthesized b y CX-chymo t ryps in-
catalyzed coupling reactions in solvent systems consisting of
buffer and ethyl acetate 3^. In comparison to a pure aqueous
medium, in which o n l y insignificant synthesis takes place,
product formation is greatly enhanced in a biphasic medi u m owing
to the extraction of the dipeptide into the organic phase.
2-5.2. Immobilized proteases
Proteases can be immobilized without loss o f function, and the
potential o f immobilized proteolytic enzymes for peptide
synthesis has been demonstrated 3 3,34^ pe p tide coupling reactions
can be carried out o n a preparative scale in which immobilized
proteases can be used with the advantage of avoiding reaction
conditions which are normally required for chemical condensation.
The simplified work-up procedure that becomes possible when
immobilized proteases are used, the long-term stability o f the
imnobilized enzyme preparations, and successful réutilisation is
2 8
-advantages in the application of covalently bound proteases can
be sunmarized as follows : the immobilized protease can easily be
recovered from the reaction mixture ; the peptides synthesized
are free from contamination b y proteolytic activities and
denatured protein ; d u e to the increased stability in the
presence of organic solvents, higher concentrations o f such
solvents can be used to influence the position o f the
thermodynamic equilibrium.
Immobilization o f chymotrypsin, trypsin and thermolysin to
various carriers has b een described T h e possibility of using
immobilized trypsin for kinetically controlled peptide bond
formation was investigated b y A. Kbnnecke et al. W ith the
serine type enzyme trypsin, excellent product yields were
obtained starting with ester carboxyl components. Covalently
immobilized trypsin catalyzed the formation o f peptide bonds with
nearly the same efficiency a s the soluble protease and could be
re-used successfully for further coupling experiments.
One of the most interesting aspect o f usi n g immobilized proteases
is the use of organic solvents as the reaction mediun. Papain
entrapped in Amberlite XAD-8 was fully active in
4-methylpentan-2-one and was used to catalyze dipeptide
-2
9-2-5.3. Polyethylene glycol modified enzymes
The hydroxyl groups o f monomethoxypolyethylene glycols
(HO-(C H2-CH2-O)nCH3) m a y be activated b y cyanuric chloride and
several other reagents a n d then coupled to lysine «-amino groups
o f proteins.
This chemical modification of proteins and enzymes with
polyethylene glycol (PEG) has become a n approach applicable to
the solution o f various problems in biological sciences. The
production of IgE caused b y protein allergens such as ovalbumin
and ragweed pollen w a s suppressed b y the treatment with
respective proteins m odified with PEG ^ . Modification of
Escherichia coli asparaginase 3®, yeast uricase ^ and snake
venom batroxobin w i t h PEG decreased their iirmunoreactivity
towards antibodies against respective proteins.
It was demonstrated that polyethylene glycol-modified enzymes
might become soluble i n organic solvents such as benzene, toluene
and chlorinated hydrocarbons, and exhibit high enzymic activities
in these organic solvents. The modified catalase catalyzed ^
decomposition of hydrogen peroxide and peroxidase catalyzed
oxidation reactions, respectively, in transparent organic
solvents. A similarly m o dified lipase was also soluble in various
organic solvents and h a d the ability to catalyze ester synthesis
3 0
-The first successful attempt to form a peptide bond b y aminolysis
in benzene was described b y Inada in 1984 ^ . Chymotrypsin was
modified in the zymogen form with 2,4-bls(0-
methoxypolyethyleneglycol)-6-chloro-s-triazine (activated PBG2),
followed by activation w i t h trypsin. The modified enzyme was
soluble in benzene and r etained its enzymic activity. Acid-amide
bond formation b y the m o d i f i e d enzyme proceeded efficiently in
benzene : benzoyl-tyrosine- (o l igo) -phenylalanine ethyl esters
were formed from N-benzoyl-L-tyrosine ethyl ester and L~
phenylalanine ethyl esters.
Since this original publication, the use o f PEG-enzymes has
dramatically increased a n d the major proteases, chymotrypsin
papain ^ , thermolysin subtilisin and trypsin ^ , have been
successfully employed for peptide synthesis in organic solvents.
In a comprehensive study o n the kinetics and specificity of
serine proteases in pep t i d e synthesis catalyzed in organic
solvents H. Gaertner a n d A. Puigserver showed that the
enzymatic synthesis obey e d Michaelis-Menten kinetics and was
consistent with a ping-pong mechanism modified b y a hydrolytic
shunt. A minimal water concentration was required for the
catalytic activity of mod i f i e d chymotrypsin in water-immiscible
solvents. However, the u s e o f PEG-modified enzymes suffers some
shortcomings including the necessity o f using hydrophobic
solvents with polar compounds, a n d the difficulty o f reisolating
3 1
-2-5.4. Reverse micelles
The solubilization of enzymes v i a reverse micelles provides a
method for the catalytic biotransformation o f water-insoluble
material. Reverse micelles are for m e d by amphiphilic molecules
(surfactants) in organic solvents ; the polar groups (heads) of
the surfactants molecules are dir e c t e d towards the interior of
the spheroidal aggregate, forming a polar core and the aliphatic
chains are directed towards the organic solvent. This is the
"reverse" of the situation in n o r m a l micelles in water. Water can
be solubilized in the polar core o f reverse micelles, forming the
water pool. The chemists are interested in reverse micelles as
versatile microreactors in w h i c h guest molecules can be brought
to reaction with novel chemical properties 5 0 . Interest from
biotechnologists has increased o v e r the last few years because
enzymes can be hosted in rev e r s e micelles without loss of
activity. The possibility o f stabilizing water-soluble enzymes
against the inactivating action o f organic solvents b y means of
surfactants has been wid e l y studied. Several enzymes,
chymotrypsin trypsin pyrophosphatase peroxidase ^
were used to demonstrate that enzymes can be entrapped into
reverse micelles formed b y s urfactants in an organic solvent. The
enzymes solubilized in this w a y ret a i n their catalytic activity
and substrate specificity.
Proteases in reverse micelles h a v e b e e n used for the synthesis o f
3 2
-synthesis o f Z-Ala-Phe-LeuNH2 starting from Z-Ala-Phe-OMe and
LeuNH2 using chymotrypsin as catalyst. Z-Ala-Phe-OMe is soluble
both in water and in isooctane, and the product is much more
soluble in isooctane than i n wate r . Ac-Phe-LeuNH2 has also been
synthesized using proteases i n reverse micelles A hollow
fiber reactor has been utilized for these peptide syntheses. This
system works well but is not su i t a b l e for preparing large amounts
of material.
More recently, Lattes ^ reported the first successful enzymatic
transesterification in a m i c e l l a r medium. A n e w type o f
microemulsion system (Aerosol-OT, water, hexanol) was used to
entrap a-chymotrypsin in a r e v e r s e micelle. N-acetyl-L-tyrosine
ethyl ester can be solubilised i n this medium to give N-acetyl-L-
tyrosine hexyl ester in a good yield.
2-5.5 Direct solubilisation o f e n z y m e s in organic solvents.
Recently, it was shown that preparations of enzymes in organic
solvents might be prepared directly. Crown ethers and cryptands
were found to be selective comple x i n g agents for a number of
proteins, allowing their d i s s o l u t i o n in non-aqueous solvents ^7.
Bovine insulin and cytochrome C were readily solubilized in
methanol b y weak complexation w i t h 18-crown-6 ether. However, the
3 3
-o f the c-omplexing agent had t-o b e used, and n -o inf-ormati-on
concerning the catalytic integrity o f the enzymes was given. A
direct application of this c o n c e p t was obtained b y the
preparation of a lipid-coated lipase w h i c h was soluble in organic
solvents such as benzene No n i o n i c and cationic lipids formed
complexs with lipases, and h i g h activity was obtained in
hydrophobic solvents.
2-5.6. Heterogeneous catalysis in an h y d r o u s organic solvents
In 1966, Dastoli first o bserved enzymatic activities of
crystalline chymotrypsin in an h y d r o u s nonpolar organic solvents
-*9. Since then, several enzymatic transformations in nonpolar
organic solvents have been reported ^0.61 _ Klibanov and Zaks have
examined the role o f water in e n z y m a t i c reactions i n a number o f
anhydrous polar and nonpolar organic m e d i a and concluded that, in
general, enzymes needed a thin l a y e r of water o n the protein
surface to retain their catalytically active conformation in
anhydrous media 60*62^ The most ade q u a t e nonaqueous media are
hydrophobic solvents that do not s t r i p the essential water from
the enzymes. Water-immiscible s o l v e n t s containing w a ter below the
solubility limit are suitable for d r y enzymes. Wit h i n this range
of water content, the enzymatic activity in a n appropriate
organic solvent can be optimized, a n d the catalysis follows
Michaelis-Menten kinetics. Recent findings indicate that the
3 4
-compounds . When water is stripped f r o m the enzyme b y a solvent,
areas of the protein which normally interact with water become
exposed. The addition o f compounds which could mimic the
interaction o f water with the protein should restore, to some
degree, enzymatic activity. The presence of 1% formamide
increased the activity o f alcohol dehydrogenase in butyl acetate
(0.4X water) 15-fold over the a c tivity in the absence o f the
additive 6 3 . Enzymatic peptide synthesis in the presence o f water
mimics, such as formamide, ethylene glycol, or methanol, was
possible using thermolysin in tert-pentyl alcohol ^ . Partial
replacement o f water with water-mimicking cosolvents was found to
be beneficial for enzymatic fragment coupling b y combining high
reaction rates and the absence o f side reactions such as
hydrolysis. This general effect w a s not limited to compounds
interacting with the protein through hydrogen bonding. Crown
ethers considerably enhanced the r a t e o f the chymotrypsin-
catalyzed transesterification o f N-acetyl-L-phenylalanine ethyl
ester with 1-propanol in n-octane It was suggested that
complexation o f the airmonium, guanidinium, and potassiun cations
at the outside o f the enzyme, by the c r own ether added, rendered
the enzyme more soluble in the apolar solvent.
Suspending enzymes in organic solvents can dramatically alter a
number of their fundamental properties. It was found that the
enzyme remembered the p H o f the last aqueous solution it has been
3 5
-corresponding ionisation states w h ich then remain both in the
solid state and in organic solvents
The substrate specificity o f chymotrypsin and subtilisin were
significantly modified by placing them in organic solvents It
was postulated that the major driving force o f substrate binding
is hydrophobic interactions between the side chain o f the amino
acid residue and the binding pocket o f the enzyme. For that
reason, the substrate specificity o f chymotrypsin in organic
solvents was reversed and there was a significant increase in
reactivity towards hydrophilic substrates.
Enzymes were found to be extremely thermostable in water-
restricted environments Whereas lipase in water at 100°C is
inactivated almost instantly, the h a l f life o f the enzyme at this
temperature in tributyrin was greater than 12 hours.
It was also reported that the lyophilisation o f chymotrypsin from
aqueous solutions containing ligands (such as N-acetyl-L-
phenylalanine) had a significant effect o n the activity o f the
enzyme in organic solvents It was postulated that the
activation was the result o f the ligand locking the enzyme in its
active conformation.
A dramatic change o f stereoselectivity was also observed b y
suspending subtilisin in anhydrous organic solvents The
enzyme readily incorporated D-amino a cid residues as donor esters
in d r y tert-pentyl alcohol. Klibanov explained this result b y
recognizing that when a substrate interacts with the enzyme,
3 6
-product ive binding of the L-ester to the active site of
subtilisin results in the release of m o r e water molecules from
the hydrophobic pocket of the enzyme than that of the D-isomer.
The process of water release is less favourable in hydrophobic
media compared with water. Thus, the re a c t i v i t y of the L-ester in
hydrophobic media decreases substantially a n d the discrimination
between the D- and L-esters is diminished.
The change in free energy during enzyme-substrate (ES)
complexation in water is considered to r e q u i r e , in addition to
other changes, the disruption of a n u m b e r o f hydrogen bonds
associated with the substrate and the e n z y m e active site ^°. A
transition from aqueous to organic solvents for the complexation
o f polar substrates with their natural e n z y m e s may result in a
tight binding o f either the substrate o r the product to the
enzyme, and a severe substrate or pro d u c t inhibition or both
could occur. This argument was used t o explain w h y several
carbohydrate-converting enzymes have b een r eported to be inactive
in organic solvents
Novel enzymatic reactions in gases and supercritical fluids have
been exploited using the heterogeneous ca t a l y s i s concept This
latter approach has the merit of facilitating the recovery o f the
products and is environmentally acceptable.
Hydrophilic organic solvents such as DMF t e n d to strip water from
enzymes and inactivate them. By site-directed mutagenesis, it was
possible to prepare a subtilisin mutant w h i c h was several hundred
3 7
-50 times more stable in dry »IF 7^. This m u t a n t was used b y C.H.
Wong for the synthesis of dipeptides 7^.
2-5.7. catalytic antibodies and artificial enzymes.
The idea o f transition state binding led t o an experimental
approach toward the design and synthesis o f immunogenic transtion
state analogs for the hydrolysis of esters a n d peptides 7 . For
application in synthesis, the most interesting developments are
the demonstrations of stereospecific transesterifications,
lipase-like hydrolysis, and aminolysis in w a t e r 7 ^.
A novel and interesting variant of enzymatic peptide synthetic
methodology was suggested by Nakajima 7 6 . H e u s e d aminoacyl-t-RNA
synthetases, enzymes which are involved i n ribosome-mediated
protein synthesis and which demonstrated h i g h specificity for
their cognate amino acids, to prepare a s e r i e s of dipeptides.
Unfortunately, this method is far from the preparative scale.
Sasaki reported the design o f an artificial catalyst for the
synthesis o f peptide bonds 7 7 . He used a c r o w n ether as scaffold
to which two thiol groups were fixed as ca t a l y t i c functions. The
educts were covalently linked b y chemical m e a n s to the enzyme
mimic via thioester bonds. Intramolecular aminolysis resulted in
the formation o f a peptide b ond with the g r o w i n g peptide still
3 8
-The peptidyltransferase, a ribosomal protein that catalyses
peptide bond formation in vivo should be a n ideal choice to serve
this function in vitro. However, its proteosynthetic activity is
dependent upon the presence of other ribosomal h e l p e r proteins,
so, when isolated from its environment, the enzyme cannot
preserve its original functions. Attempts h ave b een made to
design and synthesize synthetase mimics but i t is too early
3 9
-CHAPTER I .APPLICATIONS O F CHYMOTRYPSIN S U S P H I D E D IN ORGANIC
SOLVBTTS
1-1. PEPTIDE SYNTHESIS CATALYZED BY C H YMOTRYPSIN IN ORGANIC
SOLVENTS.
1-1.1. Introduction.
As illustrated in the introduction, great interest has been shown
in the use of proteases to catalyze peptide b o n d formation. The
drawbacks inherent to catalysis in an a q u e o u s environment
(unfavourable thermodynamic equilibrium, n a r r o w substrate
specificity, and undesirable proteolysis of the peptide) have
b een overcome b y the use o f proteases in biphasic aqueous organic
mixtures 32, reverse micelles 33 or chemically modified enzymes
in organic solvents ^ 5 . However, recently it h a s been found that
enzymes can be catalytically active in anhydrous organic solvents
and, under these conditions, show a new range o f properties, e.g.
relaxed stereospecificity and mod i f i e d substrate
specificity 62,66,79,80,81^ Although the use o f chymotrypsin as a
suspension in organic solvents for esterification ®2 and
4 0
-b een used to catalyze peptide bond formation under these
conditions.
That the internal configuration of protein m o l ecules could be
radically altered b y organic solvents has b e e n shown In
the early sixties, some interest was shown in catalytic
properties of protein crystals. Carboxypeptidase
ribonuclease-S and Chymotrypsin 87,88 have a l l been reported
to show typical catalytic activities when suspended in aqueous
sulfate solutions.
In 1966, Dastoli reported the chymotrypsin-catalyzed hydrolysis
of Ac-L-TyrOEt in methylene chloride containing 0 . 25% water ^ .
Crystals recovered from suspension in dichloromethane for up to 5
d ays showed no measurable loss in reactivity u p o n assay with
acetyl-L-tyrosine ethyl ester in aqueous solution.
B a sed on this initial observation, we postulated that, if
Chymotrypsin is able to catalyze hydrolytic reactions in organic
solvents, peptide synthesis was envisageable b y aminolysis o f
esters.
Substantial evidence indicates that chymotrypsin-catalyzed
hydrolyses proceed in two steps The first reaction is
acylation of serine-195 of the enzyme and the second is a
subsequent deacylation. Uater is not unique i n the latter
4 1
-(nucleophiles N) for example may result in changes i n the rates
and products (Scheme 1). Therefore, peptides are e x p e c t e d to be
synthesized enzymatically when amino acid derivatives, peptides
or their derivatives are used as nucleophiles N in S c h e m e 1.
Ac-Phe-X + Chymotrypsin -j££ » Ac-Phe-X. Chymotrypsin
If water is restricted in the reaction mixture, one c a n expect
high yields of peptides to be obtained
1-1.2. Materials and methods.
Enzyme.
Crystalline bovine pancreatic a -Chymotrypsin (EC 3.4.21.1)
(type II) was purchased from Sigma as a lyophilized p o w d e r with
4 2
-1.0 m o j oI o f benzoyl-L-tyrosine ethyl ester (BTEE) per m i n u t e at
p H 7.8 at 25°C). The amount o f bound water was determined b y the
Karl-Fischer method and found to be 5.4% (w/w) for t h e native
enzyme and 1.5% (w/w) for the enzyme extensively d r i e d in a
desiccator over P2O5. The enzyme was used in this s t udy without
further purification.
Amino acid derivatives, peptides and their derivatives.
Ac-L-TyrOEt, Bz-L-TyrOEt, Ac-L-PheOEt, Ac-L-TrpOEt, Bz-L-AlaOEt
were obtained from Sigma; amino acid amides were p u r c h a s e d from
Bachem AG.
Esterification of the substrates was carried out with saturated
solutions o f hydrochloric acid in the given alcohol. V a r i o u s Z-
amino acid esters were prepared b y reaction o f the e s ter w i t h N-
(benzyloxycarbonyloxy)succinimide in chloroform. BOC-derivatives
were prepared by reaction o f the ester with di-tert-butyl
dicarbonate in aqueous medium or in organic solvents. Protec t i o n
of the amino groups o f the substrates b y an acetyl g r o u p was
carried out b y reaction with neat acetic anhydride i n the
presence o f a base or b y acetyl chloride.
Z-dipeptide substrates were prepared b y reaction of the p r o tected
Z-amino acid and the amino acid ester with EEDQ i n dry
dichloromethane.
0-alanine amide, L-phenyllactic amide, and «
their BOC-derivatives according to a procedure d e v eloped by
Muramatsu
Enzymatic synthesis of peptides.
In a typical experiment, to a solution of the protected amino
acid ester and the amino acid amide (40 mM, 1:1 molar ratio) in
anhydrous dichlorome thane (distilled over calcium hydride
immediately prior to use) was added 1 mg/ml of chymotrypsin,
followed b y the addition o f 0.25% (v/v) of water. The resulting
suspension was then stirred at room temperature for a certain
period of time. In all cases investigated, the desired compound,
if formed, precipitated during the course of the reaction. The
solvent was then evaporated under reduced pressure, the residue
was thoroughly washed with water, and the product recrystallized
from hot methanol.
If hydrochloride or trifluoroacetate salts o f the a m ino acid
amides were to be used as substrates, the salt was first
4 4
-C o mputer graphics,
The computer-assisted molecular modelling systems RIMG and MOLOC
h ave b een developed at Hoffmann-LaRoche, Basel. There are built
around two novel generally applicable united-atom force field
methods, which are complementary to each other. One method uses
automatic referencing to input structures, the other is b a sed on
a small generic set o f fixed external parameters. Both modelling
systems provide interactive modelling and nonlinear optimisation
techniques in a well balanced and highly functional combination.
T hey contain algorithms for different graphic representations o f
m o l ecular structures and packing as well as efficient procedures
for structure refinement, energy evaluation, and conformation
analysis. They incorporate extensive tools for complex
geometrical as well as logical manipulations o f molecular
structures and provide facilities for fast on-line storage and
retrieval o f molecular structures and structural fragments.
The two specially designed relational data base systems R O CSD
(Roche Cambridge Structural Data Base), containing the converted
data o f the Cambridge Crystallographic Data Files, and ROPDB
(Roche Protein Structural Data Base), holding the converted d ata
o f s ome 300 protein structures from the Brookhaven Protein Data
-
45
-1-3. Results and discussion.
The procedure w a s carried out on a 0.4-4 nmol scale. No activity
was found with l ess than 0.2% of water, indicating that a minimal
amount of water is absolutely essential for enzymatic catalysis.
This essential w a t e r presumably allows the enzyme to maintain its
native conformation, as well as sufficient flexibility, and hence
to retain its catalytic activity 80.
The role o f the essential water will be extensively discussed in
Chapter II, all the literature in this area have reported the
effect of water i n terms of total water or in terms of added
water. In fact, o n e can distinguish two different states of water
in this type o f non-aqueous enzymology, i.e. the water bound to
the enzyme powder a n d the water freely dissolved in the organic
solvent. This differentiation is useful because o nly the enzyme-
bound water seems to decide its catalytic activity. Lysozyme has
been used in most studies in this area ^1. Using a variety o f
experimental techniques, it has been shown that the hydration o f
a protein consists o f a number of distinct stages : below
0.07 g/g (water p e r protein) water predominantly interacts with
charged groups. Bet w e e n 0.07 and 0.25 g/g, water forms clusters
which grow and c o v e r most of the surface. Within the range o f
hydration, for lysozyme, there is a significant increase in the
mobility o f the protein matrix. The enzymatic activity o f
lysozyme becomes detectable at as low as 0 .2 g/g (at which only
4 6
-in parallel with the prote-in's motional properties. At 0.38 g/g
(about 300 water molecules per protein molecule) the protein
molecule is fully hydrated and the enzymatic activity reaches 10%
of that in water. The amount of water necessary for an enzyme to
function in organic solvents is one o f the major questions of
non-aqueous enzymology. The fact that the distribution o f water
between the enzyme and the organic solvent governs enzymatic
activity implies that enzymes suspended in hydrophobic solvents
require substantially less water for activity than those
suspended in hydrophilic ones.
Zaks and Klibanov ^ 3 , by studying the dependence on the amount of
water bound to the enzyme, showed that in hydrophobic solvents
water tends to partition into the enzyme so that even very small
concentrations o f water in the solvent « 1 % ) yield u p to 30%
water on a protein. In a similar study concerning lipase-
catalyzed intramolecular esterification in benzene, Yamane ^2
established that the degree of hydration of enzyme molecules
necessary to exhibit full activity had a saturation level. At a
higher free water content, the enzyme powder phase contains more
water than the saturated level. In fact, the optimal water
content is a function not only of the organic solvent, but o f the
enzymes as well, a n d to a lesser extent o f the enzymatic process.
In our study, dichloromethane rapidly established itself as the
most suitable solvent. With peptide substrates, a compromise has
to be reached bet w e e n the need for a highly hydrophobic solvent
-
47
-character of the substrates. The best established solvents like
tert-pentyl alcohol and isooctane could not be used because of
the insolubility o f the substrates.
If the importance o f water for enzymatic activity is well
understood, only sparse reports have been published to explain
firstly the absence o f enzymatic activity in anhydrous solvents
which could provide valuable insights into protein structure,
folding and dynamics, and second the effect of the solvent on the
protein structure. Solid-state NMR spectroscopy was used to show
that the catalytic triad of a -lytic protease was intact when the
enzyme was suspended in acetone and isooctane ^3. Even if this
reveals that part o f the catalytic site remains intact, this
technique does not provide information about the structural
integrity o f the enzyme.
If the molecules o f solvents can diffuse into the crystal
lattice, they might then interact strongly with the structure by,
for example, disturbing hydrophobic clusters, reinforcing
hydrogen bond networks, o r directly interacting with the binding
o f the substrates. The tridimensional structure of the enzyme can
therefore be m o dified and slight changes in the conformation of
selected residues mig h t have profound consequences for the
reactivity and specificity of the enzyme.
This concept was demonstrated b y experiments carried out with V -
chymotrypsin w h ich showed that an enzyme inactive in water can
become active w h e n suspended in organic solvents due to the
4 8
-This example will be discussed in Chapter III.
In order to provide support for this hypothesis, the behaviour o f
chymotrypsin in organic solvents was investigated and by
combining the experimental results with molecular modelling, a
qualitative analysis o f the tridimensional structure was
attempted.
In n o cases was any peptide formation detected in the absence of
water. Under the aforementioned conditions, n o hydrolysis of the
final peptide was observed, indicating that the added water is
not available for hydrolysis.
T o examine specificity at the P^' position (notation of Schechter
and Berger ^ ) , experiments were performed with various amino
acid derivatives as nucleophiles. The results are sumnarized in
Table 1.
Table 1 ; Effect o f the nucleophile specificity o n synthesis.
Donor ester Nucleophile Product Yield ( X ) Reaction
time (hrs)
Ac-L-TyrOEt L-PheNH2 Ac-L-Tyr-L-PheNH2 96 6
L-LeuNH2 Ac-L-Tyr-L-LeuNH2 95 6
L-LeuOMe -
0
72" L-ValNH2 Ac-L-Tyr-L-ValNH2 92 18
" L-AlaNH2 Ac-L-Tyr-L-AlaNH2 84 18
" L-MetNH2 Ac-L-Tyr-L-Me tNH2 86 12
" L-ProNH2 -
0
72" 6-AlaNH2 -
0
72 [image:49.359.59.336.27.408.2]4 9
-Hydrophobic and b u lky amino acid amides as nucleophiles were
found to be suitable substrates for peptide synthesis, but the
imino acid derivative, L-prol inamide, was not accepted as a
substrate under any o f the conditions tested.
It is worth noting that amino acid esters (e.g. methyl L-
leucinate) were not readily accepted as nucleophiles. The
explanation for this observation was provided b y molecular
modelling experiments. Picture 1 shows a color-coded display of
putative hydrogen b o nds for Ac-L-Tyr-L-AlaNMe, covalently bound
to serine-195 (tetrahedral oxyanion intermediate) of
chymotrypsin.
5 0
-The oxyanion is well engaged in the oxyanion hole (two hydrogen
bonds to backbone NH). T h e C-terminal peptide-NH of the amide
engages in a strong hydrogen bond to the backbone-CO of
phenylalanine-41. The N-terminal peptide-CO forms an
intramolecular hydrogen b o n d to the N H group of the scissile
peptide bond.
Picture 2 shows the color-coded display o f hydrogen bonding
between Ac-L-Tyr-L-AlaOBz 1 and the active site domain of
chymotrypsin.
Picture 2
One can note the lack o f a n y hydrogen bond between the benzyl
5 1
-The loss o f one strong h ydrogen bonding interaction to the
peptide-CO of phenylalanine explains why amino acid esters are
not suitable nucleophiles. T his loss may account for a loss in
the free binding energy between 2-4 kcal/mol in water, and due to
the new organic environment w h ere hydrophobic interactions will
be diminished but hydrogen bonding greatly enhanced, the figure
might well be m uch higher.
The importance o f the hydrogen bonding between the amide moiety
a n d the backbone-CO o f Phe-41 was further demonstrated b y using
d-alaninamide as a nucleo p h i l e . /3-Alaninamide having one more
methylene unit should therefore lose this stabilising
interaction. As shown in Table 1, no reaction was observed when
fl-alaninamide was u s e d as a nucleophile.
T h e specificity o f a-chymotrypsin for hydrophobic amino acid
nucleophiles is often explained in terras o f hydrophobic
interactions between the side-chain of the nucleophile and the
peptide backbone. I f that explanation were correct, one might
then assume that b y replacing w a ter with another reaction mediun,
one would change the substrate specificity. This is not
experimentally observed as sho w n b y the results given in Table 1.
One can consider the suspended enzyme in organic solvents as
being shielded from the b ulk o f the solvent. Firstly, the enzyme-
b o und water will protect the enzyme molecule from any detrimental
effect o f the solvent. Second, the enzyme being in a quasi
crystalline state, the penetration o f the solvent into the
5 2
-Therefore, one might assune that if molecules can diffuse into
the enzyme molecule, n o direct solvation phenomenon will be
observed, but rather the solvent will coordinate with tb® enzyme
matrix at specific binding sites such as hydrophobic clusters,
isolated hydrophobic side-chains etc.
The enzyme will then still remain in a highly organized state,
the only difference with a n aqueous environment being the
presence at specific points o f solvent molecules. According to
Dewar ^ , the proper substrate o f an enzyme is generally believed
to fit its active site closely. If so, adsorption of the
substrate in the active site will necessarily lead to the
expulsion of all molecules o f solvents (i.e. water) from between
them, leaving bare substrate in contact with bare enzyme. Any
subsequent reaction between the enzyme and the adsorbed substrate
will then take place in the absence of solvent, i.e., as it would
in the gas phase, n o water separating the groups that are
directly involved in the reaction. Recent work has shown that gas
phase chemistry differs greatly from solution chemistry. In
particular, many reactions that take place slowly in solution
take place with little o r n o activation, and hence extremely
rapidly, in the gas phase. According to Dewar, this suffices to
explaining the high rates observed in enzymatic reactions. But,
more interestingly, a substrate too big to fit into the active
site o f an enzyme cannot react w i t h it, while a substrate which
is too small cannot squeeze out all the molecules of solvent.