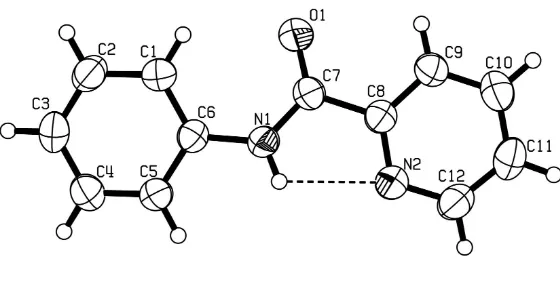
N Phenylpyridine 2 carbamide
6
0
0
Full text
Figure
Related documents
Full text
Figure
Related documents