Copyright © 1999, American Society for Microbiology. All Rights Reserved.
Genotyping of the CCR5 Chemokine Receptor by Isothermal NASBA
Amplification and Differential Probe Hybridization
JOSEPH W. ROMANO,1SURYA TETALI,2EUN MI LEE,1ROXANNE N. SHURTLIFF,1XUE PING WANG,2 SAVITA PAHWA,3MARK H. KAPLAN,2ANDCHRISTINE C. GINOCCHIO2*
Advanced BioScience Laboratories, Inc., Kensington, Maryland 20895,1and Department of Infectious Disease2and
Department of Pediatrics and Immunology,3North Shore University Hospital, Manhasset, New York 11030
Received 9 April 1999/Returned for modification 24 June 1999/Accepted 7 September 1999
The human CCR5 chemokine receptor functions as a coreceptor with CD4 for infection by macrophage-tropic isolates of human immunodeficiency virus type 1 (HIV-1). A mutated CCR5 allele which encodes a protein that does not function as a coreceptor for HIV-1 has been identified. Thus, expression of the wild-type and/or mutation allele is relevant to determining the infectibility of patient peripheral blood mononuclear cells (PBMC) and affects disease progression in vivo. We developed a qualitative CCR5 genotyping assay using NASBA, an isothermal nucleic acid amplification technology. The method involves three enzymes and two oligonucleotides and targets the CCR5 mRNA, which is expressed in PBMC at a copy number higher than 2, the number of copies of DNA present encoding the gene. The single oligonucleotide set amplifies both alleles, and genotyping is achieved by separate hybridizations of wild-type- and mutation-specific probes directly to the single-stranded RNA amplification product. Assay sensitivity and specificity were demonstrated with RNAs produced in vitro from plasmid clones bearing the DNA encoding each allele. No detectable cross-reactivity between wild-type and mutation probes was found, and 50 copies of each allele were readily detectable. Analysis of patient samples found that 20% were heterozygous and 1% were homozygous for the CCR5 mutation. Thus, NASBA is a sensitive and specific means of rapidly determining CCR5 genotype and provides several technical advantages over alternative assay systems.
The CC-CKR5 (or CCR5) cell surface molecule serves as the natural receptor for the CC (or)-chemokines (reviewed in reference 16). The CCR5 molecule has also been shown to serve as a coreceptor, along with the CD4 molecule, for the entry of macrophage-tropic, non-syncytium-inducing (i.e., pri-mary) strains of human immunodeficiency virus type 1 (HIV-1) during infection (1, 7). A series of studies have demonstrated that the CCR5 genotype is important in determining host sus-ceptibility to HIV-1 infection, as well as in determining disease progression (9, 15). For example, it was demonstrated that cells from two HIV-1-negative individuals with multiple expo-sures to HIV-1⫹patients were resistant to infection by primary
macrophage-tropic virus in vitro (12). Sequence analysis deter-mined that both of these individuals encoded the same ho-mozygous deletion mutation of 32 bp in the CCR5 gene. This deletion mutation in both alleles of the gene results in a trun-cated form of the receptor that is not detected on the cell surface. A more extensive survey of patients has revealed that approximately 3% of 612 HIV-1-exposed but antibody-nega-tive individuals were homozygous for this deletion mutation (6). In the same study, it was shown that the deletion allele occurred at a frequency of about 0.10 in the U.S. Caucasian population. There were no homozygous deletion mutations detected among more than 1,300 HIV-1⫹ patients, and the
frequency of heterozygosity for the mutation was elevated among patients surviving for more than 10 years. Interestingly, the deletion mutation appears to be of low prevalence among people of Asian or African ancestry (9). The relevance of the CCR5 receptor in HIV-1 infection is further established by the observation that the -chemokines RANTES, MIP-1␣, and
MIP-1(which naturally bind the CCR5 receptor) will block infection of macrophage-tropic isolates of HIV-1 in vitro (5). The studies which addressed the CCR5 genotype and its role in HIV-1 pathogenesis involved the use of several molecular methods for the genotyping process. In this report, we describe the use of the isothermal nucleic acid sequence-based ampli-fication (NASBA) method for CCR5 genotyping analysis. NASBA (10) is an isothermal process that is highly appropriate for the amplification of RNA. Amplification is achieved through the coordinated activities of three enzymes (avian myeloblastosis virus reverse transcriptase, RNase H, and T7 RNA polymerase) and two DNA oligonucleotides that are specific for the target sequence of interest. Amplification in NASBA is transcription based and results in the production of large amounts of single-stranded RNA that is antisense to the original target sequence. The single-stranded RNA product can then be readily detected through the hybridization of an appropriately labeled oligonucleotide DNA probe. With CCR5, a single NASBA system which amplifies both the wild-type and the mutated alleles was designed. Discrimination between the two amplification products was achieved at the detection step by means of independent hybridizations with wild-type- and mutation-specific probes. It is the abundance of the CCR5 transcript in patient peripheral blood mononuclear cells (PBMC) and the appropriateness of NASBA for RNA amplification which enabled development of this genotyping assay. The isothermal nature of the amplification procedure along with an easily detected single-stranded RNA amplifica-tion product further justifies the use of NASBA technology in genotyping analyses.
MATERIALS AND METHODS
Obtaining and processing of biological samples.After obtaining informed consent, whole-blood samples were collected in VACUTAINER EDTA antico-agulant tubes (Becton Dickinson, Franklin Lakes, N.J.) from 89 randomly
se-* Corresponding author. Mailing address: Department of Infectious Disease, North Shore University Hospital, 300 Community Dr., Manhasset, NY 11030. Phone: (516) 719-1079. Fax: (516) 719-1254. E-mail: cginocch@nshs.edu.
959
on August 17, 2020 by guest
http://cvi.asm.org/
lected HIV-1-seropositive patients attending the North Shore University Hospi-tal Center for AIDS Research and Treatment (Manhasset, N.Y.). Additional samples were collected from four human T-cell leukemia virus type 1 (HTLV-1)-positive patients and four HIV-1-seronegative persons with a history of high-risk exposure to HIV-1. Whole bloods were centrifuged for 20 min at 1,000⫻g in a swinging-bucket rotor (Spinchron R; Beckman Instruments, Fullerton, Cal-if.) within 4 h of specimen collection. PBMC were obtained from the whole blood either by centrifugation through Ficoll Hypaque or through the use of cell preparation tubes (Becton Dickinson). Resulting PBMC were either viably cryo-preserved for use later or processed directly for nucleic acid extractions. Nucleic acids were isolated from approximately 105cells (NASBA assay only) or 2⫻106
cells (NASBA and PCR assays) by the guanidine isothiocyanate-acidified silica procedure of Boom et al. (3). After isolation, 5 to 10% of the nucleic acid extract was used in the NASBA reactions and PCRs. Alternatively, to obtain total RNA, the method of Chomczynski and Sacchi (4) was used.
Amplification by NASBA of CCR5 transcript RNA. Amplification by the NASBA technique was achieved by a modified version of the procedure of Romano et al. (18). Briefly, the 20-l amplification reaction mixture contained 5 l of the nucleic acid extract material in a solution containing 40 mM Tris (pH 8.5); 5 mM dithiothreitol; 12 mM MgCl2; 70 mM KCl; 2.0 mM (each) ATP, CTP,
and UTP; 1.5 mM GTP; 0.5 mM ITP; 1.0 mM (each) dATP, dCTP, dGTP, and dTTP; 0.1g of bovine serum albumin perl; 0.1 U of RNase H; 40 U of T7 RNA polymerase; 8 U of avian myeloblastosis virus reverse transcriptase; 15% dimethyl sulfoxide; and 0.2M (each) oligonucleotides (P1 and P2). Two sets of oligonucleotides, specific for CCR5 amplification, were synthesized. Design of these oligonucleotides was based on the reported sequence for the CCR5 gene (GenBank accession no. U57840), and the oligonucleotides were as follows: P1A, 5⬘ AATTCTAATACGACTCACTATAGGGAGCAGCGGCAGGACCAGCC CCA 3⬘; P1B, 5⬘AATTCTAATACGACTCACTATAGGGAGAGATTCCCGAG TAGCAGATGACCATGA 3⬘; P2A, 5⬘GGCTGTGTTTGCGTCTCTCCCA 3⬘; and P2B, 5⬘ TTTGGGGTGGTGACAAGTGTGATCA 3⬘. The italicized se-quences of the P1 oligonucleotides (A and B) indicate the overhang portion encoding the T7 RNA polymerase promoter. All four P1-P2 oligonucleotide combinations were evaluated for performance in the NASBA system.
Hybridization analysis of CCR5 NASBA products.The portion of the CCR5 gene targeted by the specified oligonucleotides spans positions 430 through 650 (the A of the ATG methionine is base 1). The probe for the wild-type amplifi-cation product (5⬘AGTATCAATTCTGGAAGAATTTCCA 3⬘, used in elec-trochemiluminescence [ECL] or32P-based detection) anneals to the positions
corresponding to bases 557 to 581, which are within the 32-base deleted portion of the mutated allele. The Ru2⫹-labeled probe specific for the mutated allele corresponds to base positions 546 to 553 and 586 to 593; however, the probe is a continuous oligonucleotide corresponding to these regions. The sequence of this mutation-specific ECL detector probe is 5⬘TCCATACA:TTAAAGAT 3⬘ (the colon signifies the junction site). Therefore, this probe should hybridize only to the mutated form of the gene, where base 553 is immediately adjacent to base 586 due to the deletion. Similarly, the probe used for32P-based detection of the
mutated allele (5⬘ TCATTTTCCATACA:TTAAAGATAGTCAT 3⬘) corre-sponds to positions 540 to 553:586 to 599. Figure 1 illustrates the position of the CCR5 NASBA primers and probes. NASBA products were analyzed by probe hybridization in one of two ways. In method 1, amplification products were vacuum transferred to nylon membranes in 2⫻SSC (1⫻SSC is 0.15 M NaCl plus 0.015 M sodium citrate) buffer with a slot blot apparatus or after electrophoretic resolution through agarose gels followed by vacuum transfer. After transfer, the blots were hybridized with probes that were 5⬘end labeled with32P by standard
methods. After hybridization (16 to 20 h), blots were washed three to four times with 1⫻SSC–1% sodium dodecyl sulfate at 50°C and exposed in autoradiogra-phy. In method 2, the alternative detection strategy involved the use of an ECL detection system (2). In this system, the wild-type and mutation probes are labeled with ruthenium (Ru2⫹), the compound responsible for the ECL signal. Amplification products were first diluted by a factor of 10 to 40 and hybridized separately in liquid phase with Ru2⫹-labeled versions of either the wild-type or the mutation detector probe. Included in these liquid hybridization reaction mixtures was a capture probe (5⬘AAAGAAGGTCTTCATTACACCTGCAGC 3⬘, positions 511 to 537) which was specific for a conserved sequence present in both wild-type and mutated allele amplification products. This capture probe was labeled at the 5⬘end with biotin and immobilized onto the surface of a strepta-vidin-coated magnetic bead (type 280; Dynal, Inc., Lake Success, N.Y.). This liquid hybridization involving the diluted NASBA reaction product, the immo-bilized capture probe, and either the wild-type or mutated Ru2⫹-labeled probe was conducted at 60°C for 5 min and then for 20 to 30 min at 41°C. Results were obtained by analysis of the hybridization reaction products in the NASBA QR System (Organon Teknika, Durham, N.C.), an ECL reader.
Cloning of the wild-type and mutated CCR5 alleles.High-molecular-weight genomic DNA was purified from pooled buffy coat PBMC by standard methods. The DNA was subjected to PCR amplification (30 cycles by standard methods) with the following primer pair: a sense primer, 5⬘ATGGATTATCAAGTGTC AAGTCCAATCTAT 3⬘, and an antisense primer, 5⬘TCACAAGCCCACAGA TATTTCCTGCTC 3⬘. The resulting 1,059-bp PCR product was ligated directly into plasmid pCR2.1 (Invitrogen, Inc., Carlsbad, Calif.) and used to transform Escherichia coliINV␣F⬘. Probes specific for the CCR5 insert were used to screen the resulting transformants, and independent clones for each allele were
ob-tained. Sequences of the clones were determined through a commercial service (Lark Technologies, Houston, Tex.). These clones were used to produce RNA corresponding to the wild-type and mutated CCR5 alleles by means of a Ribo-probe in vitro transcription kit (Promega Corp., Madison, Wis.). After DNase treatment of the in vitro transcription products and purification with RNeasy (Qiagen, Inc., Santa, Clarita, Calif.), quantitation of this RNA was achieved by measuring the optical density at 260 nm in triplicate and using the mean value to calculate the copy number.
PCR amplification of CCR5 DNA.DNA PCR determination of CCR5 geno-type was performed according to the previously described method (8). Briefly, the PCRs were performed in 50-l volumes, each comprising 5l of nucleic acids, 5l of 10⫻PCR buffer (100 mM tris-HCl [pH 8.3], 500 mM KCl, 15 mM MgCl2, 0.01% [wt/vol] gelatin), 0.2 mM each deoxynucleoside triphosphate, 50
pmol each of the sense (5⬘CAATGTGTCAACTCTTGACAGG 3⬘) and anti-sense (5⬘ACCTGCATAGCTTGGTCCAACC 3⬘) primers, 33.5l of distilled water, and 0.5l ofAmpli Taqenzyme (Perkin-Elmer Corp., Foster City, Calif.). The DNAs in the reaction mixtures were denatured for 1 min at 94°C on a thermal cycler (PCR 9600 system; Perkin-Elmer Corp.) and subjected to 35 cycles of PCR amplification. Each cycle consisted of denaturation at 94°C for 30 s, followed by annealing at 58° for 30 s and then extension at 72°C for 30 s. Following the 35 cycles of PCR there was an extension for 10 min at 72°C.
Electrophoresis and detection of PCR-amplified products.Following PCR, the CCR5 amplified DNAs were electrophoretically separated on a 2% agarose gel stained with ethidium bromide. DNA fragments were visualized by UV scanning with a UVP gel documentation system (Integral Technologies, Inc.). Samples homozygous for the wild-type CCR5 allele demonstrate a single 547-bp ampli-con, samples homozygous for the mutated (32-bp deletion) CCR5 allele dem-onstrate a single 515-bp amplicon, and samples with heterozygous CCR5 alleles demonstrate two DNA bands at 547 and 515 bp.
RESULTS
Basic assay performance.A schematic representation of the CCR5 genotyping NASBA assay is provided in Fig. 1. The approach was to conduct a single NASBA of RNA obtained from patient PBMC and subject the resulting amplicons to independent hybridization analyses with probes that are spe-cific for either the wild-type or the mutated allele. Multiple oligonucleotides for CCR5 amplification by NASBA were de-signed such that there were two antisense P1 sequences down-stream from the 32-base deletion mutation site and two sense P2 sequences upstream from this region (map positions 602 to 623 for P1A, 626 to 650 for P1B, 465 to 486 for P2A, and 430 to 454 for P2B). The wild-type-allele-specific probe was posi-FIG. 1. Strategy for genotyping at the CCR5 locus by NASBA. The wild-type CCR5 allele contains a 32-base sequence (black bar) which is deleted from the mutated allele. The oligonucleotides used for amplification by NASBA of the CCR5 mRNA were designed as shown with two antisense P1 oligonucleotides (A and B) downstream from the deletion site and two sense P2 oligonucleotides (A and B) upstream from the deletion site. Map positions are provided in Materials and Methods. All four combinations were used in initial amplification reaction evaluations. Resulting amplicons were screened by hybridization analysis with either the wild-type probe (WT), which is specific for the sequence that is deleted from the mutated allele, or with the mutation-specific probe (Mut), which strad-dles the resulting junction region created by the deletion. The capture probe, used in ECL detection of amplification products, is located immediately down-stream from P2A (not shown). The P1A-P2B primer combination was used in all subsequent experiments.
on August 17, 2020 by guest
http://cvi.asm.org/
tioned in the 32-base region that is present only in the wild-type gene. The mutation-specific probe corresponded to the junction region that is created by the deletion event.
All four possible combinations of sense and antisense oligo-nucleotides were used to test the amplification by NASBA of in vitro-produced wild-type CCR5 RNA. The NASBA products were analyzed with the 32P-5⬘-end-labeled CCR5 wild-type probe in a blot-based hybridization analysis (method 1 in Ma-terials and Methods), and the resulting autoradiograms were evaluated. Although all four oligonucleotide combinations were functional, it was found that the P1A-P2B combination produced the maximum amount of NASBA product (data not shown). Further, it was determined that a KCl concentration of 70 mM was optimal for amplification with the P1A-P2B primer set. Therefore, all subsequent NASBA reactions were per-formed with these primers in 70 mM KCl.
Sensitivity of the NASBA assay.Several strategies were em-ployed to evaluate the sensitivity of the NASBA assay for CCR5 mRNA. Initially, it was determined that the assay could detect the CCR5 target mRNA sequence in as little as 50 pg of total RNA obtained from PBMC. In the standard NASBA reaction conducted with PBMC extracts obtained by the method of Boom et al. (3), CCR5 mRNA was readily detected with input material equivalent to 5,000 cells. However, a more informative evaluation of sensitivity requires the use of RNA produced in vitro that corresponds to the CCR5 target se-quence. To obtain this RNA, PCR was conducted with genomic DNA that had been extracted from pooled human PBMC with primers that spanned the entire coding region of the CCR5 gene. Since the CCR5 gene does not contain in-trons, the DNA sequence is contiguous with the transcribed RNA (19). Further, the use of DNA extracted from pooled PBMC enhances the likelihood of both wild-type and mutated forms of the gene being amplified in the PCR. The resulting PCR product was ligated directly into the TA cloning vector pCR2.1, and bacterial transformants were screened with a 32P-labeled capture probe (see Materials and Methods) which hybridizes to both wild-type and mutated clones. Through ad-ditional screening steps, independent clones harboring the
wild-type and mutated CCR5 alleles were isolated and the identities of these clones were confirmed by means of DNA sequence analysis. The specific wild-type and mutated CCR5 plasmids were then used to produce separate batches of both forms of RNA by means of in vitro transcription.
NASBA reactions were performed with independent titra-tion dilutitra-tions of the wild-type and mutated CCR5 in vitro-produced RNA products. Both forms of RNA were amplified with the same oligonucleotide set (i.e., P1A and P2B); how-ever, amplification products were diagnosed separately by hy-bridization with ruthenium-labeled versions of either the wild-type or the mutation probe. Also included in this hybridization reaction mixture was a capture probe which has sequence com-mon to both alleles and is immobilized on the surface of a magnetic bead. Thus, the hybridization reaction product con-sists of the NASBA amplicons annealed to the capture probe on the bead surface, with the allele-specific ruthenium-labeled detector probe being bound at an independent position of the amplicons. These hybridization products are then evaluated in an ECL reader that quantifies the amount of light induced during the ECL process (2). The amount of light is a function of the quantity of amplicon that is bound to both the magnetic bead and the detector probe. Thus, results of the hybridization analysis were determined by automated quantitation of the resulting ECL signal with the NASBA QR system.
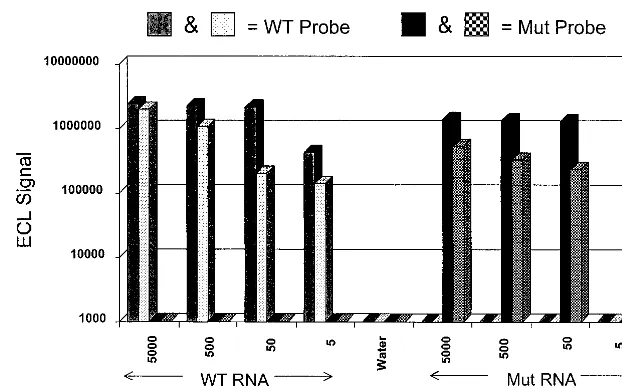
It was found that the assay exhibited approximately equal sen-sitivities for both forms of the CCR5 RNA. The assay detected 50 copies of in vitro-produced wild-type and mutated RNAs in du-plicate attempts, as presented in Fig. 2. Although the results presented in Fig. 2 indicate that 5-copy sensitivity is possible, the inability to detect this amount of template in both trials of mu-tated RNA establishes a 50-copy limit of sensitivity for the assay. Further, it was found that there was no cross-reactivity between the wild-type- and mutation-specific probes. Results of these sen-sitivity and specificity experiments are summarized in Fig. 2. Im-portantly, the CCR5 sequence is highly homologous to the se-quence of an independent chemokine receptor, CCR2 (GenBank accession no. U80924). However, the CCR5 NASBA assay failed to detect 5⫻105copies of in vitro-produced RNA corresponding FIG. 2. Sensitivity and specificity of the CCR5 NASBA assay. Different input quantities (5 to 5,000 copies) of wild-type (WT) CCR5 RNA and RNA corresponding to the mutated allele (Mut) that were produced in vitro were amplified with the P1A-P2B oligonucleotide set. Detection of the amplification products was achieved with the ECL system described in Materials and Methods. Each input RNA was amplified in duplicate, and each reaction mixture was tested with both the WT- and Mut-specific probes. Results show that the assay is capable of at least 50-copy sensitivity (WT and Mut) and that there is no cross-reactivity between the specific probes on the alternative RNAs.
on August 17, 2020 by guest
http://cvi.asm.org/
to the CCR2 transcript (data not shown). However, this CCR2 RNA was readily detected by an NASBA assay specific for CCR2 transcripts.
Analysis of patient PBMC samples.After the initial evalu-ation of the NASBA-based CCR5 assay, the system was ap-plied to the analysis of patient PBMC samples. Nucleic acids were obtained by the silica-based extraction method of Boom et al. (3), which leads to the isolation of both RNA and DNA. However, the isothermal nature of NASBA renders the am-plification method specific for the RNA versions of the targets; there is no opportunity for a double-stranded DNA molecule to denature. Therefore, there is no contribution to the NASBA product from the background DNA obtained from the cells. After amplification as described earlier, reaction products were subjected to both32P-labeled probe detection and ECL-based hybridization analysis. For the32P-labeled probe detec-tion system, duplicate 5-l aliquots of the NASBA reaction product were either resolved through independent agarose gels and then vacuum transferred to nylon membranes or vac-uum transferred directly as slot blots. After independent hy-bridizations with the allele-specific probes, the genotypes of individual patients could be determined by scoring for the presence or absence of bands on the resulting autoradiograms. A homozygous wild-type genotype was indicated by the pres-ence of a band only on the wild-type probe autoradiogram; the homozygous mutation genotype was indicated by the presence of a band only on the mutation probe autoradiogram. Het-erozygotes had bands on both autoradiograms. Results from an analysis of eight patients are summarized in Fig. 3. Evalu-ation of these autoradiograms indicated that patients 2, 3, 4, and 8 were homozygous for the wild-type allele and that pa-tients 1, 5, 6, and 7 were heterozygous at the CCR5 locus.
Patient samples were also analyzed by means of the ECL-based detection system. After the NASBA process was applied, resulting amplicons from each patient were subjected to inde-pendent liquid-phase hybridization analyses with the wild-type-and the mutation-specific ruthenium-labeled probes. These
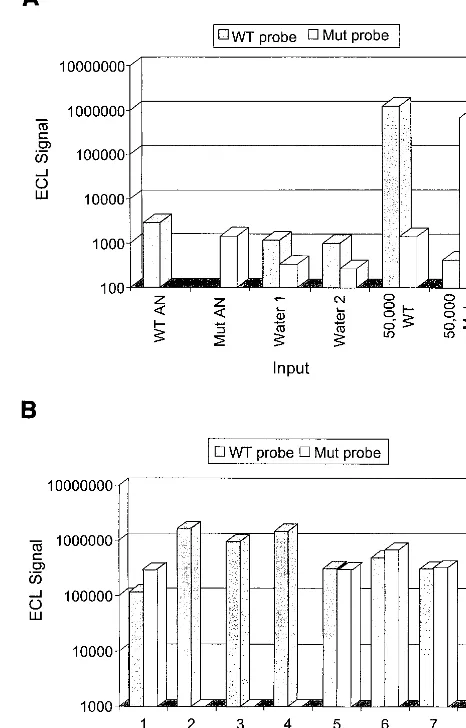
hy-bridization products were then analyzed in the NASBA QR System ECL reader. After appropriate subtraction of back-ground signals, results were scored as positive or negative for the wild-type and mutation probes based on the resulting ECL signal. Figure 4 summarizes the results obtained from the same eight patients represented in Fig. 3. Figure 4A depicts the ECL results for the control materials used during the patient anal-ysis. Clearly, the ECL-based assay is specific, with no cross-reactivity between wild-type- and mutation-specific probes. Furthermore, all of the negative control reactions were scored at background ECL levels. In Fig. 4B, results obtained from the analysis of patient PBMC are provided. This ECL-based version of the assay again demonstrated that patients 2, 3, 4, and 8 had the homozygous wild-type CCR5 genotype and that patients 1, 5, 6, and 7 had the heterozygous genotype.
The specificity of the NASBA-ECL assay was further
vali-FIG. 3. CCR5 NASBA genotyping analysis of patient PBMC by32
P-labeled-probe detection. Approximately 5% of the nucleic acid extracts obtained from patient PBMCs were amplified with the P1A-P2B CCR5 NASBA assay oligo-nucleotide set. The reaction products were resolved in duplicate agarose gels and then vacuum transferred to nylon membranes. Each membrane was hybridized with a32P-labeled version of either the wild type (upper blot)- or mutation (lower
blot)-specific CCR5 probes. Lanes 1 to 8 contain patient PBMC samples; lanes 9 and 10 contain negative (water) controls; lanes WT contain 105copies of
wild-type CCR5 RNA; and lanes Mut contain 105copies of mutated CCR5
RNA.
FIG. 4. CCR5 NASBA genotyping analysis of patient PBMC by ECL detec-tion. (A) Analysis of control samples. Assay-negative (AN) ECL signals were obtained for Ru2⫹versions of the wild-type (WT)- and mutation (Mut)-specific
probes by hybridization with a magnetic-bead-immobilized capture probe and without amplicon material. Water-only controls were also amplified in duplicate and subjected to analysis with the WT and Mut probes. ECL signals for these controls were equivalent to background levels. Positive-control reaction mixtures consisted of 50,000 copies of in vitro-produced WT RNA or 50,000 copies of in vitro-produced Mut RNA. (B) Analysis of patient PBMC samples. Amplification products obtained for the same eight patients whose results are depicted in Fig. 3 were subjected to ECL-based detection analysis by independent hybridization with the WT and Mut probes.
on August 17, 2020 by guest
http://cvi.asm.org/
dated by comparing the CCR5 genotype results obtained by the NASBA assay with results obtained from the analysis of identical samples by a DNA PCR assay (8). Initially, nucleic acids extracted from 105 PBMC were used for both the NASBA and DNA PCR assays. Although the CCR5 genotype was readily determined by the NASBA assay for all samples, several samples were not amplified sufficiently by PCR to de-termine the genotype (data not shown). Subsequently, all com-parative studies were performed with an input volume of 2⫻
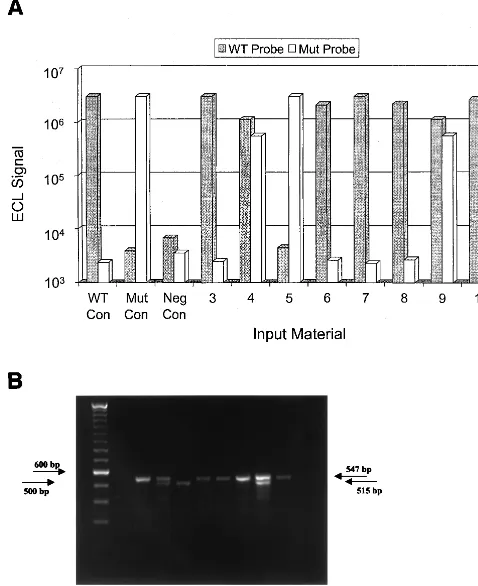
106PBMC. The NASBA and DNA PCR genotype results of 61 patients tested were 100% concordant. An example of com-parison results identifying all three genotypic profiles is shown in Fig. 5, with panel A depicting NASBA results and panel B depicting DNA PCR results obtained for eight patients.
A summary of the CCR5 NASBA genotyping analysis of multiple patient PBMC extracts is provided in Table 1. The populations analyzed included 79 HIV-1⫹patients, 10 HIV-1⫹
long-term survivors, 4 HTLV-1⫹patients, and 4
HIV-1-sero-negative patients with a history of high-risk exposure to HIV-1 through intravenous drug use and/or heterosexual contact. In total, 97 patients were analyzed; 79% were homozygous for the wild-type gene, 20% were heterozygous, and 1% were homozy-gous for the mutation. The results demonstrate that this NASBA-based assay can readily be applied to patient material for the purpose of genotyping at the CCR5 locus. In all cases where samples were analyzed by both the32P- and ECL-based detection methods, there was 100% concordance between the results. Therefore, the convenience and speed of the ECL-based method dictates its use in any laboratory setting.
DISCUSSION
The CCR5 gene encodes a cell surface chemokine receptor molecule that also functions as a coreceptor for macrophage-tropic isolates of HIV-1. Importantly, a naturally occurring mutated allele for the CCR5 gene which encodes a 32-base deletion has been identified. This mutated allele produces a truncated form of the protein that is not expressed on the cell surface. Consequently, PBMC from patients who are homozy-gous for the deletion mutation cannot be infected with mac-rophage-tropic virus and appear to be resistant to infection in vivo despite multiple exposures (12). Heterozygosity at this gene apparently correlates with a slower progression of HIV-1-related disease (6, 8, 9). Consequently, patient genotyping at this locus is important in assessing prognosis.
We have developed a rapid, sensitive, and specific genotyping assay for the human CCR5 gene. The assay utilizes isothermal amplification by NASBA of CCR5 RNA obtained from patient PBMC. Although detection and actual sequence typing of the resulting amplicons was achievable with radiolabeled probes in blot-based hybridization analysis, a technically more favorable ECL-based detection method was developed. This noniso-topic, liquid hybridization detection method involves rutheni-um-labeled probes that are specific for amplicons generated from the wild-type and mutated alleles of CCR5. Hybridization results are evaluated in this system with an automated ECL reader, allowing for rapid diagnosis of NASBA products. This assay system has several advantages over other amplification-based methods which can be applied in genotyping. First, it is isothermal and does not require specialized equipment for amplification. The nucleic acids are obtained from the clinical material by the extraction method of Boom et al., which can be applied to a wide range of sample types. Second, the product of the NASBA amplification process is single-stranded RNA, which is readily analyzed by probe hybridization without the need for a denaturing step. Genotyping assays directed at DNA typically depend on the presence of only two copies of target material per cell. The NASBA assay targets RNA and therefore exploits the presence of a more abundant target material in cells where the gene is expressed.
FIG. 5. CCR5 genotyping by NASBA and DNA PCR. (A) NASBA products generated from the PBMC of eight patients were subjected to ECL-based de-tection analysis by independent hybridization with wild-type (WT)- and mutation (Mut)-specific probes. Bars WT Con, wild-type control samples produced signals with only the WT probe; bars Mut Con, mutation control samples produced signals with only the Mut probe; bars Neg Con, negative-control samples had undetectable signals with both probes. The PBMC of patients 3, 6, 7, 8, and 10 (bars so numbered) produced positive signals with the WT probe only, indicating a homozygous wild-type genotype. The PBMC of patients 4 and 9 produced signals with both the WT and Mut probes, indicating a heterozygous (wild-type/ 32-bp-deletion) genotype. The PBMC of patient 5 produced a positive signal with the Mut probe only, indicating a homozygous 32-bp-deletion genotype. (B) CCR5 PCR amplification products generated from PBMC from the same eight patients whose results are shown in panel A were visualized by ethidium bromide staining after electrophoresis on a 2% agarose gel. Three different amplification patterns were observed. Lane 1, DNA size marker; lane 2, negative water PCR control (no bands visualized); lanes 3, 6, 7, 8, and 10, homozygous wild-type amplicons bp band); lanes 4 and 9, heterozygous deletion amplicons (547-and 515-bp b(547-ands); lane 5, homozygous deletion amplicons (515-bp b(547-and).
TABLE 1. CCR5 genotyping analysis of patient PBMC by NASBA
Patient statusa No. of patients with PBMC CCR5 genotype b:
WT/WT WT/Mut Mut/Mut Total
HIV-1⫹ 65 14 0 79
HIV-1⫹LTS 6 4 0 10
HIV-1⫺HRE 3 0 1 4
HTLV-1⫹ 3 1 0 4
aLTS, long-term survivors; HRE, patients subjected to high-risk exposure. bWT, wild-type allele; Mut, mutation allele. Seventy-nine percent of all pa-tients were homozygous for the wild-type allele, 20% were heterozygous, and 1% were homozygous for the 32-bp deletion.
on August 17, 2020 by guest
http://cvi.asm.org/
Importantly, when the NASBA system was applied to clini-cal specimens (i.e., patient PBMC), the frequency of the mu-tated allele was found to be similar to what had been previously reported (23). The frequency of heterozygotes among individ-uals of western European heritage is approximately 20%. The NASBA-measured frequency in this study was also 20%. Only one individual (with a history of intravenous drug abuse) who was homozygous for the mutated allele was identified in this study. This person is the spouse of an HIV-1-infected individ-ual and has remained HIV-1 seronegative despite frequent high-risk exposure through the sharing of needles with other HIV-1-infected individuals and through spousal heterosexual contact. Similarly, several other investigators have identified persons with the homozygous 32-bp deletion genotype who are resistant to HIV-1 infection (6, 9, 12, 17, 20–23). The fact that only one individual with the homozygous mutated genotype was found in this study is consistent with the facts that nearly all of the patients tested were HIV-1 positive and that the frequency of homozygosity in the general population is only 1% (12). Interestingly, three persons with a history of multiple exposure to HIV-1 through intravenous drug abuse, but not infected with HIV-1, demonstrated homozygous wild-type CCR5 genotypes. Additional studies are in progress to deter-mine the nature of HIV-1 infection resistance in these persons. Interestingly, the CCR5 NASBA genotyping assay has been used in parallel with a PCR assay directed against CCR5 DNA, and the results obtained with the two assays were 100% con-cordant. An advantage of the NASBA assay was the ability to perform CCR5 genotyping on a significantly lower number of PBMC than was required for the DNA PCR assay (1⫻ 105 [NASBA] versus 2⫻106[PCR]). Although it may be possible to reconfigure the DNA PCR assay for greater sensitivity by means of an alternative detection method, the results pre-sented here indicate greater sensitivity by the NASBA assay targeting CCR5 mRNA. Furthermore, NASBA CCR5 geno-typing can be successfully performed directly on whole-blood samples, eliminating the time-consuming process of PBMC isolation (21a). Thus, NASBA technology provides a rapid, simple, and specific means for accurate genotype determina-tion at the CCR5 locus. The clinical significance of this marker in the prognosis of HIV-1-infected patients and the ease with which this genotyping assay can be applied suggest that appli-cation of the NASBA assay on a wide scale is appropriate. Further, the NASBA assay provides an effective means for conducting population demographic studies. Moreover, NASBA technology can be applied to the detection of other relevant polymorphisms identified in the CCR5 gene (11, 13, 14), perhaps in a multiplex format. In summary, targeting the CCR5 RNA transcript with the NASBA assay system is an effective method for determining the genotype at this locus.
ACKNOWLEDGMENT
This study was funded in part by the Jane and Dayton Brown and Dayton Brown, Jr., Molecular Diagnostics Laboratory at North Shore– LIJ Health System Laboratory, Lake Success, N.Y.
REFERENCES
1.Alkhatib, G., C. Combadiere, C. C. Broder, Y. Feng, P. E. Kennedy, P. M. Murphy, and E. A. Berger.1996. CC-CKR5: a RANTES, MIP-1␣, MIP-1 receptor as a fusion cofactor for macrophage-tropic HIV-1. Science272: 1955–1958.
2.Blackburn, G., H. Shah, J. Kenten, J. Leland, R. A. Ramin, J. Link, J. Peterman, M. J. Powell, A. Shah, D. B. Talley, S. K. Tyagi, E. Wilkins, T. G. Wu, and R. J. Massey.1991. Electrochemiluminescence detection for devel-opment of immunoassays and DNA probe assays for clinical diagnostics. Clin. Chem.37:1534–1539.
3.Boom, R., C. J. Sol, M. M. Salisman, C. L. Jansen, P. M. E. Wertheim-van Dillen, and J. van der Noordaa.1990. Rapid and simple method for purifi-cation of nucleic acids. J. Clin. Microbiol.28:495–503.
4.Chomczynski, P., and N. Sacchi.1987. Single step method of RNA isolation by acid gaunidinium thiocyanate-phenol-chloroform extraction. Anal. Bio-chem.162:156–159.
5.Cocchi, F., A. L. DeVico, A. Garzino-Demo, S. K. Arya, R. C. Gallo, and P. Lusso.1995. Identification of RANTES, MIP-1␣, and MIP-1as the major HIV-suppressive factors produced by CD8⫹T cells. Science270:1811–1815. 6.Dean, M., M. Carrington, C. Winkler, G. A. Huttley, M. W. Smith, R. Allikments, J. J. Goedert, S. P. Buchbinder, E. Vittinghoff, E. Gomperts, S. Donfield, D. Vlahov, R. Kaslow, A. Saah, L. Rinaldo, R. Detels, and S. J. O’Brien.1996. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter He-mophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 273:1856–1862.
7.Dragic, T., V. Litwin, G. P. Allaway, S. R. Martin, Y. Huang, K. A. Na-gashima, C. Cayanan, P. J. Maddon, R. A. Koup, J. P. Moore, and W. A. Paxton.1996. HIV-1 entry into CD4⫹cells is mediated by the chemokine receptor CC-CKR5. Nature381:667–673.
8.Eugen-Olsen, J., A. K. Iversen, P. Garred, U. Koppelhus, C. Pedersen, T. L. Benfield, A. M. Sorensen, T. Katzenstein, E. Dickmeiss, J. Gerstoft, P. Skinhoj, A. Svejgaard, J. O. Nielsen, and B. Hofmann.1997. Heterozygosity for a deletion in the CKR-5 gene leads to prolonged AIDS-free survival and slower CD4 T-cell decline in a cohort of HIV-seropositive individuals. AIDS 11:305–310.
9.Huang, Y., W. A. Paxton, S. M. Wolinsky, A. U. Neumann, L. Zhang, T. He, S. Kang, D. Ceradini, Z. Jun, K. Yazdanbakhsh, K. Kunstman, D. Erickson, E. Dragon, N. R. Landau, J. Phair, D. D. Ho, and R. A. Koup.1996. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat. Med.2:1240–1243.
10. Kievits, T., B. van Gemen, D. van Strijp, R. Schukkink, M. Dircks, H. Adriaanse, L. Malek, R. Sooknanan, and P. Lens.1991. NASBA isothermal enzymatic in vitro nucleic acid amplification optimized for the diagnosis of HIV-1 infection. J. Virol. Methods35:273–286.
11. Kostrikis, L. G., Y. Huang, J. P. Moore, S. Wolinsky, L. Zhang, Y. Guo, J. Phaie, A. U. Neumann, and D. D. Ho.1998. A chemokine receptor CCR2 allele delays HIV-1 disease progression and is associated with a CCR5 promoter mutation. Nat. Med.4:350–353.
12. Liu, R., W. A. Paxton, S. Choe, D. Ceradini, S. Martin, R. Horuk, M. E. MacDonald, H. Stuhlmann, R. A. Koup, and N. R. Landau.1996. Homozy-gous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell86:367–377.
13. Martin, M. P., M. Dean, M. W. Smith, C. Winkler, B. Gerrard, N. L. Micheal, B. Lee, R. W. Doms, J. Margolick, S. Buchbinder, J. J. Goedert, T. R. O’Brien, M. W. Hilgartner, D. Vlahov, S. J. O’Brien, and M. Car-rington.1998. Genetic alteration of AIDS progression by a promoter variant of CCR5. Science282:1907–1911.
14. McDermott, D. H., P. A. Zimmerman, F. Guignard, C. A. Kleeberger, S. F. Leitman, and P. M. Murphy.1998. CCR5 promoter polymorphism and HIV-1 disease progression: Multicenter AIDS Cohort Study (MACS). Lan-cet352:866–870.
15. McNicholl, J. M., D. K. Smith, S. H. Qari, and T. Hodge.1997. Host genes and HIV: the role of chemokine receptor gene CCR5 and its allele. Emerg. Infect. Dis.3:261–271.
16. Premack, B. A., and T. J. Schall.1996. Chemokine receptors: gateways to inflammation and infection. Nat. Med.2:1174–1178.
17. Quillent, C., E. Oberlin, J. Braun, D. Rousset, G. Gonzalez-Canali, P. Me-tais, L. Montagnier, J. L. Virelizier, F. Arenzana-Seisdedos, and A. Beretta. 1998. HIV-1 resistance phenotype conferred by combination of two separate inherited mutations of the CCR5 gene. Lancet351:14–18.
18. Romano, J. W., R. N. Shurtliff, M. G. Sarngadharan, and R. Pal.1995. Detection of HIV-1 infection in vitro using NASBA: an isothermal RNA amplification technique. J. Virol. Methods54:109–119.
19. Samson, M., O. Labbe, C. Mollereau, G. Vassart, and M. Parmentier.1996. Molecular cloning and functional expression of a new human CC-chemokine receptor gene. Biochemistry35:3362–3367.
20. Samson, M., F. Libert, B. Doranz, J. Rucker, C. Liesnard, C. M. Farber, S. Saragosti, C. Lapoumerouilie, J. Cognaux, C. Forceille, G. Muyldermans, C. Verhofstede, G. Burtonboy, M. George, T. Imai, S. Rana, Y. Yi, R. J. Smyth, R. G. Collman, R. W. Doms, G. Vassart, and M. Parmentier.1996. Resis-tance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature382:722–725.
21. Smith, M. W., M. Dean, M. Carrington, C. Winkler, G. A. Huttley, D. A. Lomb, J. J. Goedert, T. R. O’Brien, L. P. Jacobson, R. Kaslow, S. Buch-binder, E. Vittinghoff, D. Vlahov, K. Hoots, M. W. Hilgartner, and S. J. O’Brien.1997. Contrasting genetic influence of CCR2 and CCR5 variants on HIV-1 infection and disease progression. Hemophilia Growth and Develop-ment Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter
on August 17, 2020 by guest
http://cvi.asm.org/
Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC), ALIVE Study. Science277:959–965.
21a.Tetali, S.Unpublished data.
22. Wilkinson, D. A., E. A. Operskalski, M. P. Busch, J. W. Mosley, and R. A. Koup.1998. A 32-bp deletion within the CCR5 locus protects against trans-mission of parenterally acquired human immunodeficiency virus but does not affect progression to AIDS defining illness. J. Infect. Dis.178:1163–1166.
23. Zimmerman, P. A., A. Buckler-White, G. Alkhatib, T. Spalding, J. Kubofcik, C. Combadiere, D. Weissman, O. Cohen, A. Rubbert, G. Lam, M. Vac-carezza, P. E. Kennedy, V. Kumaraswami, J. V. Giorgi, R. Detels, J. Hunter, M. Chopek, E. A. Berger, A. S. Fauci, T. B. Nutman, and P. M. Murphy. 1997. Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk. Mol. Med.3:23–36.