ANALYSIS OF A-TO-IRNAEDITING IN SCHIZOPHRENIA RISK GENE MIR137
Harish Pudukodu
Honors Thesis ENHS
Department of Environmental Sciences and Engineering
Gillings School of Global Public Health
The University of North Carolina at Chapel Hill
December 7th, 2016
Approved:
________________________
Dr. James J. Crowley, Department of Genetics
________________________
Dr. Louise M. Ball, Department of Environmental Sciences and Engineering
ABSTRACT
Schizophrenia is a complex psychiatric disorder that likely emerges as a result of
interacting genetic and environmental risk factors. A prominent hypothesis for the etiology of
schizophrenia is that schizophrenia emerges as a result of pre- and/or perinatal viral
infection-induced inflammation in genetically predisposed individuals. This gene-environment interaction
may be mediated by genetic regulators such as microRNA 137 (miR137), a regulatory RNA with
a set of related SNPs that is highly associated with schizophrenia, and epigenetic phenomena
such as RNA editing, which has been previously associated with schizophrenia and infection.
Here I interrogate this gene-environment interaction by quantifying RNA editing of miR137,
assessing the functional impact of this editing, and predicting its effects on schizophrenia risk in
conjunction with a risk SNP. High-throughput RNA sequencing coupled to the PrimerID
methodology revealed that miR137 is edited at a statistically significant level (p < 0.05) in both
fetal human brain and adult mouse brain. Site-directed mutagenesis and luciferase assays
demonstrated that A-to-I modification of certain pertinent bases in miR137 impart a large
decrease in affinity to a target sequence. Finally, mathematical modeling and simulations based
on empirical findings suggest that this RNA editing and a MIR137 schizophrenia-risk SNP
significantly modulate inflammation-based schizophrenia risk burden (p < 2 x 10-16). These
findings demonstrate that infection, the MIR137 gene, and RNA editing likely interact to
promote schizophrenia risk, and thus this study serves to elucidate the cooperative influences of
ACKNOWLEDGEMENTS
I would like to thank Dr. Shuntai Zhou, Bryant Su, Dr. Kensuke Sakamoto, Dr. Ron
Swanstrom, Dr. Louise Ball, and Dr. James Crowley for their contributions to this work. This
research was supported by K01MH094406 (PI: Dr. James Crowley). This research was funded in
part by a Summer Undergraduate Research Fellowship from the Office of Undergraduate
Research at the University of North Carolina at Chapel Hill.
TABLE OF CONTENTS
Abstract ... 2
Acknowledgements ... 3
Table of Contents ... 3
List of Tables and Figures... 4
Introduction ... 5
Materials and Methods ... 7
Computational RNA Analyses ... 7
RNA Preparation and Sequencing ... 8
Mutagenesis, Luciferase, and RT-PCR Experiments ... 9
Mathematical Modeling of MIR137 System Dynamics ... 10
Statistical Analyses ... 11
Results ... 11
Adult mouse and fetal human brains exhibit statistically significant levels of A-to-I
MIR137 editing ... 12
A-to-I editing of MIR137 significantly changes target affinity ... 13
MIR137 SNP rs1625579 modulates inflammatory burden in a manner significantly dependent on miR137 A-to-I editing ... 15
Discussion and Conclusions ... 16
MIR137 is subjected to A-to-I RNA editing... 16
A-to-I RNA editing of MIR137 changes target affinity ... 17
Schizophrenia risk burden from infection is modulated by a SNP in MIR137 and miR137 editing rate ... 18
References ... 19
Tables and Figures ... 24
Appendix ... 32
Supplementary Tables and Figures ... 32
Supplementary Computational Methods... 35
LIST OF TABLES AND FIGURES Tables and Figures ... 24
Figure 1 ... 24
Table 1b ... 25
Figure 2 ... 26
Figure 3 ... 27
Figure 4 ... 28
Figure 5 ... 29
Figure 6 ... 30
Figure 7 ... 31
Appendix ... 32
Supplementary Table 1 ... 32
Supplementary Figure 1 ... 33
Supplementary Figure 2 ... 35
INTRODUCTION
The Psychiatric Genetics Consortium released a landmark paper in 2014, which identified
numerous risk loci that are associated with schizophrenia[1]. One of the most highly associated
haplotypes is positioned around the MIR137 locus, which codes for a microRNA that targets
other schizophrenia-associated genes such as CACNA1C and TCF4[1-42]. This discovery has
prompted considerable functional work to determine the effects of various key MIR137 single
nucleotide polymorphisms (SNPs) on the expression of miR137 and downstream targets[2, 3, 5,
43]. This work has led to the identification of gene-expression effects resulting from MIR137
schizophrenia-associated targets[2, 3, 5, 43]. This suggests that MIR137 plays a central role in the etiology of
schizophrenia.
It has been discovered in recent years that microRNAs are subject to A-to-I editing by the
various forms of the adenosine deaminase acting on RNA (ADAR) enzyme[44-52]. This
phenomenon, which causes specific adenosine bases in double stranded RNA to be converted to
inosine, has been determined to have functional impacts on microRNA biogenesis and
subsequent targeting, even leading to altered sets of target genes for a given microRNA[44-51].
In prior high-throughput RNA sequencing experiments, our group has identified possible
non-background levels of A-to-I editing in the miR137 sequence (data not shown). I hypothesize that
such A-to-I editing of miR137 and/or the interaction between the ADAR enzymes and miR137
may be involved in the etiology of schizophrenia.
The phenomenon of RNA editing has actually been associated with schizophrenia in a
number of ways[53-55]. For example, the age of onset of schizophrenia is associated with
expression levels of the ADAR1-A isoform and expression levels of multiple ADAR isoforms
are significantly altered in dorsolateral prefrontal cortex tissue samples from patients with
schizophrenia[53]. Of note is the fact that ADAR1-A is the interferon-inducible isoform of
ADAR1, which offers an interesting connection to the viral hypothesis of schizophrenia[56-65].
This hypothesis suggests that schizophrenia may develop in genetically predisposed individuals
following an intense immune response from the mother, likely triggered by a viral infection,
while the individual is still in the womb[56-65]. The altered expression of ADAR1-A in the brain
of patients with schizophrenia, coupled to its interferon-inducible (and thus infection-inducible),
lends support to the viral hypothesis of schizophrenia and simultaneously offers an etiological
Apart from ADAR1-A’s standard RNA editing role in the nucleus, it also acts as an
inducing agent for Dicer, an important enzyme in the biogenesis pathway of miRNAs[66, 67].
More specifically, ADAR1-A is capable of increasing Dicer’s Vmax four-fold, and thus enables
Dicer to produce mature miRNAs at a faster rate[66]. Interestingly, it seems that ADAR1-A
preferentially performs the latter cytoplasmic function because of its tendency to localize to the
cytoplasm in greater proportion compared to other ADAR forms[68].
Given the facts that miR137 may experience A-to-I RNA editing, miR137 almost
certainly interacts with ADAR1-A via Dicer, and ADAR1-A is induced by interferon (which is
released during viral infection), I hypothesize that infection-mediated interferon release and
schizophrenia-associated SNPs in MIR137 interact with ADAR1-A via A-to-I editing of miR137
and Dicer-mediated biogenesis to influence schizophrenia risk. This hypothesis can be parsed
into three specific aims: (1) to determine if miR137 is subjected to A-to-I RNA editing in fetal
human and adult mouse brain, (2) to determine if such editing has any functional consequences
in vitro, and (3) to determine if a schizophrenia-associated MIR137 SNP modulates marginal
schizophrenia risk changes due to infection-mediated interferon release and subsequent
ADAR1-A induction. I tested the first specific aim using a high-throughput RNADAR1-A sequencing
methodology called PrimerID[69, 70], I tested the second specific aim using site-directed
mutagenesis and luciferase assay data, and I tested the third specific aim using a predictive
modeling approach implemented in MATLAB.
MATERIALS AND METHODS
Computational RNA Analyses
molecules[71]. In short, the algorithm has been trained on hADAR1/dsRNA reaction data to
assign editing likelihood scores at adenosine bases in a custom dsRNA sequence based on nearby
ribonucleotide identities[71]. The sequence for the human precursor miR137 molecule was found
using the UCSC Genome Browser[72] and this sequence was entered into the InosinePredict web
application to produce A-to-I edit prediction data.
Secondary structure prediction for pre-miR137 was performed using RNAstructure, which
utilizes thermodynamic models and nearest neighbor parameters to predict RNA secondary
structure[73]. Visual output from this software was analyzed systematically to identify plausible
and implausible sites of A-to-I RNA editing. Implausible sites were deemed to be all unpaired
bases in the miR137 secondary structure and plausible sites were deemed to be paired adenosine
bases in the miR137 secondary structure.
RNA Preparation and Sequencing
We used a previously described protocol to construct Primer ID MiSeq sequencing
libraries[70]. In brief, cDNA was made using primers containing a block of random nucleotides
(Primer ID). The cDNA primer sequences were 5’-
GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNTAGCTGCCGCTG
GTACTCTCCTCGA -3’ and 5’-
GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNTACCTGCCGCTG
GTACTCTCCTCGA -3’ for human and mouse sample, respectively. After purification, cDNA
was amplified using two rounds of PCR and MiSeq indexed primers were incorporated. The
primer sequence of the first round PCR forward primer was 5’-
GGTGA-3’ for both human and mouse samples. Purified and pooled libraries were sequenced
using Illumina MiSeq 150 bp pair-end sequencing. We used the Illumina bcl2fastq pipeline
(v.1.8.4) for the initial processing of sequencing data, followed by the previously described
Primer ID consensus pipeline[70] to create template consensus sequences (available at
https://github.com/SwanstromLab/PID). We analyzed mutations at each nucleotide positions of
the template consensus sequences.
Mutagenesis, Luciferase, and RT-PCR Experiments
Plasmid constructs for each combination of edit were created using site-directed
mutagenesis and standard cloning procedures. For the luciferase assays, Hek293 cells were split
into a 96 well plate with 3*104 cells per well with 100 µL of media per well. Approximately 18 hours after splitting, a mixture of pLuc137TRGTx4, Renilla, optiMEM, Lipofectamine 2000, and
the desired mutagenic sequence (or the wild variation or the control pCMVPL) was made and
incubated for 20-30 minutes. After incubation, these mixtures were added to each of the wells.
This left triplicate wells for each transfected DNA sequence. After 2 days of growth, the media
was dumped, the cells were was once, and then lysed passively for 15 minutes. Finally, a
dual-luciferase assay system was used to measure both Firefly and Renilla Luciferase levels.
For the quantitative RT-PCR assays, Hek293 were split into 3 12 well plates with 2*105 cells per well and 1 mL of media per well. 28 hours later, the mutagenic and control plasmids
were mixed with lipofectamine 2000 and incubated for 20-30 minutes. These mixtures were
randomly added to wells of each of the plates, leaving triplicate wells for each transfected DNA
sequence again. After 3 days, Trizol Reagent was added to each of the wells and RNA was
purified RNA using a NCode™ VILO™ miRNA cDNA Synthesis Kit. Finally, some of these
cDNA was used to run and RT-PCR using a SYBR Green Master Mix.
Mathematical Modeling of MIR137 System Dynamics
The temporal dynamics of interferon (IFN), ADAR1-A, miR137 (MIR), TCF4 mRNA
(TM) and protein (TP), dendritic cells (DC), and inflammation burden (IB) in response to an
infection were modeled using a system of coupled ordinary differential equations (ODEs)
implemented in MATLAB. This modeling system encapsulated the interactions among these
relevant physiological entities, as they relate to inflammation, through standard biochemical
kinetics paradigms such as mass-action kinetics and Michaelis-Menten kinetics. Infection was
modeled as entrance of pathogen, which incorporated a custom function to specify square-wave
input.
The ODEs used in this system are as follows:
dPathogen
dt = SquareWave(t, pathogen period, pathogen length, pathogen high, pathogen low) − (pathogen clearance rate) ∗ Pathogen
dIFN
dt = (IFN release parameter) ∗ Pathogen ∗ DC − (IFN degradation rate) ∗ IFN
dADAR1A
dt = (ADAR1 − A synthesis parameter) ∗ IFN − (ADAR1 − A degradation rate) ∗ ADAR1A
dMIR
dt = (basal miR137 transcription rate) ∗ (1 − 0.3 ∗ (binary SNP presence)) ∗ (1 + 2.98 ∗ ( ADAR1A
20 )) − (miR137 degradation rate) ∗ MIR
E = Proportion of MIR that is edited = (basal editing rate) ∗ (1 +0.6 ∗ ADAR1A ADAR1B )
dTM
dt = (basal TM transcription rate) − (TM degradation rate) ∗ TM −
0.16 ∗ E ∗ MIR ∗ TM ∗ (Targeting Vmax) (Targeting Km) + TM
−(1 − E) ∗ MIR ∗ TM ∗ (Targeting Vmax) (Targeting Km) + TM
dTP dt =
TM ∗ (Protein Vmax) (Protein Km) + TM
−TP ∗ (Degradation Vmax) (Degradation Km) + TP
dDC
dt = (basal DC production rate) + (TCF4 stimulation factor) ∗ TP − (DC degradation rate) ∗ DC
dIB
dt = (inflammation factor) ∗ IFN
Sensitivities to parameters were calculated via one-by-one variation of parameter values
Statistical Analyses
Edit frequency significance testing was performed in Microsoft Excel, haplotype
frequency analyses were performed in MATLAB, luciferase and RT-PCR data analyses were
performed in JMP and R, and statistical analyses of the mathematical modeling results were
performed in R. Edit and haplotype frequency analyses employed the Poisson distribution to
model probabilities of individual edit events, while the remaining statistical analyses utilized
standard Gaussian procedures.
RESULTS
Pre-miR137 is a predicted target of hADAR1
Based on consensus sequence data derived from preliminary, exploratory high-throughput
RNAseq experiments performed by our group, we hypothesized that the precursor miR137
molecule would experience A-to-I RNA editing at three sites: Bases 29, 69, and 74 (Figure 1).
To initially assess the biochemical and physical plausibility of this potential editing activity, we
utilized a published hADAR1 activity prediction tool known as InosinePredict[71].
InosinePredict uses predictive algorithms developed from hADAR1/dsRNA reaction data to
assign a score to each base of a queried dsRNA molecule, reflecting the likelihood of A-to-I
editing by hADAR1[71]. We analyzed the likelihood of editing of MIR137 using the pre-miR137
sequence fed into the InosinePredict tool.
The InosinePredict algorithm predicted that many, but not all, of the adenosine bases in the
precursor miR137 molecule would be edited at a significant level by hADAR1 (Figure 1a). Of
note, the three initially hypothesized editing sites in pre-miR137 were among the high likelihood
Because the InosinePredict tool assumes a perfectly paired dsRNA molecule, the predicted
A-to-I editing sites were assessed for structural feasibility of editing. To this end, we used a
published web server-based software known as RNAstructure to predict the secondary structure
of pre-miR137[73]. Secondary structure prediction via RNAstructure revealed that some of the
predicted hADAR1 sites are not biologically plausible A-to-I editing sites because those bases
are not paired in a double-stranded manner (Figure 1b). These implausible sites of predicted
editing were ignored in downstream analyses.
Adult mouse and fetal human brains exhibit statistically significant levels of A-to-I MIR137
editing
Following our computational interrogation of the plausibility and likelihood of A-to-I
editing in pre-miR137, we sought to determine if the phenomenon is truly exhibited in brain
tissue from adult mice and fetal humans. To accomplish this, we utilized a published method
called PrimerID[70] in conjunction with high-throughput RNA sequencing. PrimerID enables
one to parse out the contribution to nucleotide-level read variability from original biological
variation (as opposed to errors introduced from library preparation and sequencing steps) using
randomized transcript barcodes[70], and thus this system allowed us to determine the true
frequency of A-to-I editing in our samples.
Whole RNA samples from adult mouse brain and fetal human brain tissues were used for
library preparation and subsequent sequencing. Read results from high-throughput RNAseq were
aligned to mouse and human genomes (depending on the sample origin) and consensus reads
were constructed. Base-level changes were statistically analyzed for significance using a Poisson
of sites, many of which corresponded directly with predicted locations, were identified as
significant A-to-I editing sites in both adult mouse brain and fetal human brain (Supplementary
Table 1). Among these sites, the highest rates of editing were determined to be at the
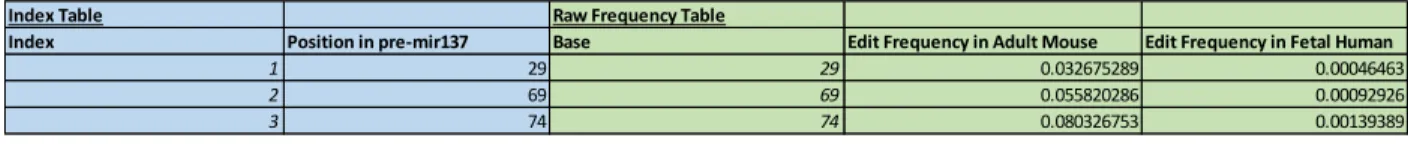
hypothesized editing sites (positions 29, 69, and 74) in both mouse and human (Table 1a).
To interrogate the potential interdependence of A-to-I editing at the high frequency sites in
fetal human and adult mouse, haplotypes and associated frequencies were determined from the
sequence read data. A MATLAB script was written to 1) predict the frequencies of edit
combinations based on the assumption of independent editing at each site, 2) calculate the true
frequencies of edit combinations from haplotype data, and 3) calculate fold differences between
predicted and observed frequencies. This analysis revealed that RNA editing at each high
frequency site was only loosely coupled in adult mouse brain and was uncoupled in fetal human
brain (Table 1b).
Of the significant A-to-I editing sites identified from the analysis of the RNAseq data, we
were interested in identifying the sites that are conserved across mouse and human for enriched
editing frequency. To this end, editing frequencies were normalized across the two test species
and compared. Of the twelve total significant editing sites identified across both species, only
editing at positions 69 and 74 (two of the initially hypothesized editing sites) were conserved and
enriched in both fetal human brain and adult mouse brain (Figure 2). Both of these positions in
the precursor miR137 molecule are present in the mature miRNA.
A-to-I editing of MIR137 significantly changes target affinity
To determine possible functional consequences of A-to-I editing of pre-miR137 on
combinations of the three edits (WT, a29g, a69g, a74g, a29g+a69g, a29g+a74g, a69g+a74g, all
three a-to-g) were made using site-directed mutagenesis in otherwise identical miR137
expression constructs. Subsequently, a luciferase assay system was designed to determine
binding activity by each of the miR137 forms. A reporter plasmid was constructed that included
a luciferase expression cassette coupled to multiple repeats of the antisense sequence of mature
miR137.
Each of the miR137 constructs (along with a positive and negative control) was tested
using this luciferase assay system, where miRNA binding to the antisense repeats would lead to
reduced luciferase activity. All of the miR137 edited forms, except for the single a74g form, led
to increased luciferase activity compared to the WT form of miR137 (Figure 3). This outcome
could be caused either by correlated differences in expression of miR137 from each construct or
by truly biological differences in targeting behavior caused by RNA editing.
To test the possibility that the luciferase data could be explained by expression
differences, each of the luciferase experiments were coupled to RT-PCR experiments to
determine expression levels of each construct. Regression of the luciferase data by the RT-PCR
data revealed that the luciferase data were not significantly associated with the RT-PCR
expression data (Supplementary Figure 1). As such, the increased luciferase activities observed
for the majority of the edited forms of miR137 are not due to differences in expression levels of
each construct. This suggests that the modification of A-to-I in select positions of miR137
MIR137 SNP rs1625579 modulates inflammatory burden in a manner significantly dependent
on miR137 A-to-I editing
To investigate the role of miR137 A-to-I editing in the potential gene-environment
interaction system posited by the viral hypothesis of schizophrenia, a mathematical modeling
framework was constructed (Figure 4) and simulated using MATLAB. This modeling
framework consisted of a set of coupled nonlinear ordinary differential equations (ODEs), which
were solved using MATLAB’s ode45 ODE solver. The parameterization of the model allowed
for the modification of production/elimination rates, binding affinities, the basal editing rate, and
presence of a single well-studied MIR137 SNP: rs1625579, which has been shown to reduce
miR137 expression[2, 43].
Schizophrenia risk was modeled as inflammatory burden, which was calculated as the
time-integral of interferon (IFN) level. This methodology is supported by numerous findings that
indicate that inflammation and dysregulated general immune response are likely etiological
factors for schizophrenia[56, 58, 61-65]. IFN release was modeled using brain dendritic cells,
which are targets of TCF4-mediated induction[25]. These inclusions, along with the other model
structures, resulted in biologically plausible dynamics for the various species of interest (e.g.
miR137 and TCF4 protein levels) (Supplementary Figure 2).
To test the influence of the SNP on inflammatory burden (and thus schizophrenia risk),
the model was simulated with and without the presence of the risk SNP under a periodic
infection spike input (Figure 5). These simulations revealed that the presence of the SNP causes
larger marginal changes in inflammatory burden in response to infection, which accumulate over
time (Figure 5). To assess whether this influence of the risk SNP on inflammatory burden
the risk SNP and with different basal RNA editing rates under a near-continuous infection input.
For each RNA editing rate tested, an inflammatory burden fold change was calculated
(inflammatory burden with the SNP over the inflammatory burden without the SNP). These fold
changes were analyzed via linear regression against the various RNA editing rate values, and this
analysis revealed that the basal RNA editing rate significantly affects the inflammatory burden
fold change due to the SNP (p < 2 x 10-16) (Figure 6). This indicates that the phenomenon of miR137 A-to-I RNA editing plays a significant role in schizophrenia risk upon infection.
DISCUSSION AND CONCLUSIONS
MIR137 is subjected to A-to-I RNA editing
To test the hypothesis that miR137 is subjected to A-to-I RNA editing by an ADAR
enzyme (likely ADAR1 due to its prevalence in the brain[44, 46, 47, 49, 66, 71, 74, 75]), the
precursor to mature miR137 (pre-miR137) was analyzed using predictive bioinformatics and the
editing rate was estimated using high throughput RNA-sequencing and PrimerID[69, 70]. The
computational analyses suggested that certain bases of pre-miR137 were likely to be subjected to
A-to-I RNA editing by ADAR1 at a significant rate (Figure 1) and the RNA-sequencing results
recapitulated these predictions in both fetal human brain and adult mouse brain (Table 1). Upon
further analysis of these RNA-sequencing data, it was revealed that the sites with the highest
relative editing rates in adult mouse brain are conserved in fetal human brain and that these sites
are present in the final mature miR137 (Figure 2), which suggests that these conserved edits
have functional consequences. All of this evidence taken together leads to the conclusion that
there is a high likelihood that miR137 is subjected to A-to-I editing in vivo.
increase with neural development[76] and the fetal human brain tissue used in this study was
inferior in quality to the adult mouse brain tissue used in this study. Given that primates typically
exhibit higher RNA editing rates than nonprimates[77], one would expect that high quality adult
human brain tissue would present with higher rates of miR137 A-to-I editing. Further molecular
transcriptomic work must be done to test this prediction and determine the true rates of miR137
A-to-I editing in adult human brains.
A-to-I RNA editing of MIR137 changes target affinity
Given the likelihood of in vivo A-to-I editing of miR137, we were interested in
determining whether such editing would have functional consequences with respect to miR137
targeting affinity. To address this question, miR137 constructs with point mutations (induced by
site directed mutagenesis) corresponding to edit combinations were assayed for antisense target
binding activity in a custom luciferase reporter-based system. This set of experiments revealed
that many of the A-to-I (A-to-G) edit combinations led to lower binding affinity between
miR137 and the antisense target (Figure 3).
The results of the above assay suggest that miR137 A-to-I editing has meaningful
functional consequences in vivo in the form of altered target affinity and subsequent changes in
gene expression networks, but further research must be done to confirm this finding and
elucidate its mechanistic nature. Given that this assay employed antisense repeats as
pseudo-targets, it may still be plausible that miR137 targeting capacity may be unchanged in vivo with
respect to real target sequences. To address this concern, future work must employ real target
sequences (derived from real target mRNAs) to better assess binding affinity changes.
consequences of miR137 RNA editing (at the biochemical and cellular levels). That being said,
the presence of this luciferase reporter assay-based in vitro data coupled to the previously
discussed evidence for editing conservation suggests that there is a true in vivo consequence to
the miR137 A-to-I editing identified in this study.
Schizophrenia risk burden from infection is modulated by a SNP in MIR137 and miR137
editing rate
Because of the IFN-inducible nature of ADAR1[44-50, 66, 71, 77], the association
between RNA editing and ADAR levels with schizophrenia[53, 70], the strong association
between MIR137 SNPs and schizophrenia, and the likely interaction between ADARs and
miR137 discovered in this study, it is likely that miR137 (and its associated genetic variation)
and RNA editing would play central roles in the development of schizophrenia risk from viral
infection, as posited by the viral hypothesis of schizophrenia. Furthermore, the role of RNA
editing in schizophrenia etiology is likely two-fold because of ADAR1’s ability to both edit
double stranded RNA molecules and promote Dicer activity[66]. To test the prediction that
miR137 A-to-I editing and MIR137 genetic variation (implemented here as the rs1625579 SNP)
interact to influence schizophrenia risk upon viral infection, a mathematical modeling framework
based on literature-derived biochemical kinetics and genetics data was constructed and
simulated. These computational experiments confirmed the aforementioned prediction: presence
of the rs1625579 SNP considerably modified inflammatory burden (Figure 5) and miR137
editing significantly influenced this SNP effect (Figure 6).
These findings lend credence to the viral hypothesis of schizophrenia and offer a
must be done to solidify the connections between viral infections, miR137 A-to-I editing, and
schizophrenia. Schizophrenia is a highly complicated and polygenic disease that almost certainly
arises from varied gene-environment interactions[1]. Given this inherent mass of complexity, it
is unlikely that any one study will provide more than a fraction of the information necessary to
understand schizophrenia. That being said, the findings in the present study related to the
influence of gene-environment interactions on schizophrenia risk allow for the development of a
basic schizophrenia risk framework, which would assist public health specialists and healthcare
practitioners as they work to prevent and treat this debilitating disorder (Figure 7). The present
study suggests that schizophrenia risk arises from genetic risk elements, environmental risk
elements, and the interaction among those genetic and environmental factors (Figure 7). Further
integrative and interdisciplinary work is crucial for the development of a quantitatively precise
model of schizophrenia risk, but the conceptual model posited here would likely be a useful
guiding framework for subsequent model development.
REFERENCES
1. Biological insights from 108 schizophrenia-associated genetic loci. Nature, 2014. 511(7510): p. 421-7.
2. Guella, I., et al., Analysis of miR-137 expression and rs1625579 in dorsolateral prefrontal cortex. J Psychiatr Res, 2013. 47(9): p. 1215-21.
3. Wright, C., et al., Potential Impact of miR-137 and Its Targets in Schizophrenia. Front Genet, 2013. 4: p. 58.
4. Genome-wide association study identifies five new schizophrenia loci. Nat Genet, 2011. 43(10): p. 969-76.
5. Collins, A.L., et al., Transcriptional targets of the schizophrenia risk gene MIR137. Transl Psychiatry, 2014. 4: p. e404.
6. Cousijn, H., et al., No effect of schizophrenia risk genes MIR137, TCF4, and ZNF804A on macroscopic brain structure. Schizophr Res, 2014. 159(2-3): p. 329-32.
8. Devanna, P. and S.C. Vernes, A direct molecular link between the autism candidate gene RORa and the schizophrenia candidate MIR137. Sci Rep, 2014. 4: p. 3994.
9. Duan, J., et al., A rare functional noncoding variant at the GWAS-implicated
MIR137/MIR2682 locus might confer risk to schizophrenia and bipolar disorder. Am J Hum Genet, 2014. 95(6): p. 744-53.
10. Egawa, J., et al., Resequencing and association analysis of MIR137 with schizophrenia in a Japanese population. Psychiatry Clin Neurosci, 2013. 67(4): p. 277-9.
11. Gianfrancesco, O., et al., Identification and Potential Regulatory Properties of Evolutionary Conserved Regions (ECRs) at the Schizophrenia-Associated MIR137 Locus. J Mol Neurosci, 2016. 60(2): p. 239-47.
12. Gonzalez-Giraldo, Y., R.E. Gonzalez-Reyes, and D.A. Forero, A functional variant in
MIR137, a candidate gene for schizophrenia, affects Stroop test performance in young adults. Psychiatry Res, 2016. 236: p. 202-5.
13. Green, M.J., et al., Genome-wide supported variant MIR137 and severe negative symptoms predict membership of an impaired cognitive subtype of schizophrenia. Mol Psychiatry, 2013. 18(7): p. 774-80.
14. Guan, F., et al., MIR137 gene and target gene CACNA1C of miR-137 contribute to schizophrenia susceptibility in Han Chinese. Schizophr Res, 2014. 152(1): p. 97-104. 15. Han, J., A. Sarkar, and F.H. Gage, MIR137: big impacts from small changes. Nat Neurosci,
2015. 18(7): p. 931-3.
16. Hill, M.J., et al., Transcriptional consequences of schizophrenia candidate miR-137 manipulation in human neural progenitor cells. Schizophr Res, 2014. 153(1-3): p. 225-30. 17. Kelly, S., et al., Genome-wide schizophrenia variant at MIR137 does not impact white matter
microstructure in healthy participants. Neurosci Lett, 2014. 574: p. 6-10.
18. Kuswanto, C.N., et al., The impact of genome wide supported microRNA-137 (MIR137) risk variants on frontal and striatal white matter integrity, neurocognitive functioning, and
negative symptoms in schizophrenia. Am J Med Genet B Neuropsychiatr Genet, 2015. 168B(5): p. 317-26.
19. Lett, T.A., et al., The genome-wide supported microRNA-137 variant predicts phenotypic heterogeneity within schizophrenia. Mol Psychiatry, 2013. 18(4): p. 443-50.
20. Li, Z., et al., Loci with genome-wide associations with schizophrenia in the Han Chinese population. Br J Psychiatry, 2015. 207(6): p. 490-4.
21. Liu, B., et al., The impact of MIR137 on dorsolateral prefrontal-hippocampal functional connectivity in healthy subjects. Neuropsychopharmacology, 2014. 39(9): p. 2153-60. 22. Morton, S.E., et al., Testing the Validity of Taxonic Schizotypy Using Genetic and
Environmental Risk Variables. Schizophr Bull, 2016.
23. Mothersill, O., et al., Effects of MIR137 on fronto-amygdala functional connectivity. Neuroimage, 2014. 90: p. 189-95.
25. Navarrete, K., et al., TCF4 (e2-2; ITF2): a schizophrenia-associated gene with pleiotropic effects on human disease. Am J Med Genet B Neuropsychiatr Genet, 2013. 162B(1): p. 1-16. 26. Ohi, K., et al., The impact of the genome-wide supported variant in the cyclin M2 gene on
gray matter morphology in schizophrenia. Behav Brain Funct, 2013. 9: p. 40.
27. Olde Loohuis, N.F., et al., The schizophrenia risk gene MIR137 acts as a hippocampal gene network node orchestrating the expression of genes relevant to nervous system development and function. Prog Neuropsychopharmacol Biol Psychiatry, 2016.
28. Patel, V.S., et al., MIR137HG risk variant rs1625579 genotype is related to corpus callosum volume in schizophrenia. Neurosci Lett, 2015. 602: p. 44-9.
29. Pu, X. and X. Xiao, No evidence of an association between MIR137 rs1625579 and
schizophrenia in Asians: a meta-analysis in 30 843 individuals. Psychiatr Genet, 2016. 26(5): p. 203-10.
30. Sanders, A.R., et al., Transcriptome study of differential expression in schizophrenia. Hum Mol Genet, 2013. 22(24): p. 5001-14.
31. Siegert, S., et al., The schizophrenia risk gene product miR-137 alters presynaptic plasticity. Nat Neurosci, 2015. 18(7): p. 1008-16.
32. Strazisar, M., et al., MIR137 variants identified in psychiatric patients affect synaptogenesis and neuronal transmission gene sets. Mol Psychiatry, 2015. 20(4): p. 472-81.
33. Sun, Y.J., et al., Association between single nucleotide polymorphisms in MiR219-1 and MiR137 and susceptibility to schizophrenia in a Chinese population. FEBS Open Bio, 2015. 5: p. 774-8.
34. Vaidyanathan, U., et al., Heritability and molecular genetic basis of antisaccade eye tracking error rate: a genome-wide association study. Psychophysiology, 2014. 51(12): p. 1272-84. 35. van Erp, T.G., et al., Schizophrenia miR-137 locus risk genotype is associated with
dorsolateral prefrontal cortex hyperactivation. Biol Psychiatry, 2014. 75(5): p. 398-405. 36. Warburton, A., et al., A GWAS SNP for Schizophrenia Is Linked to the Internal MIR137
Promoter and Supports Differential Allele-Specific Expression. Schizophr Bull, 2016. 42(4): p. 1003-8.
37. Warburton, A., et al., Characterization of a REST-Regulated Internal Promoter in the Schizophrenia Genome-Wide Associated Gene MIR137. Schizophr Bull, 2015. 41(3): p. 698-707.
38. Whalley, H.C., et al., Impact of a microRNA MIR137 susceptibility variant on brain function in people at high genetic risk of schizophrenia or bipolar disorder.
Neuropsychopharmacology, 2012. 37(12): p. 2720-9.
39. Wright, C., et al., Meta gene set enrichment analyses link miR-137-regulated pathways with schizophrenia risk. Front Genet, 2015. 6: p. 147.
41. Xia, S., et al., Experimental validation of candidate schizophrenia gene CALN1 as a target for microRNA-137. Neurosci Lett, 2015. 602: p. 110-4.
42. Xiao, X., et al., Evaluation of European Schizophrenia GWAS Loci in Asian Populations via Comprehensive Meta-Analyses. Mol Neurobiol, 2016.
43. Warburton, A., et al., A GWAS SNP for Schizophrenia Is Linked to the Internal MIR137 Promoter and Supports Differential Allele-Specific Expression. Schizophr Bull, 2015. 44. Bazak, L., et al., A-to-I RNA editing occurs at over a hundred million genomic sites, located
in a majority of human genes. Genome Res, 2014. 24(3): p. 365-76.
45. Cui, Y., T. Huang, and X. Zhang, RNA editing of microRNA prevents RNA-induced silencing complex recognition of target mRNA. Open Biol, 2015. 5(12).
46. Dupuis, D.E. and S. Maas, MiRNA editing. Methods Mol Biol, 2010. 667: p. 267-79. 47. Keegan, L.P., A. Gallo, and M.A. O'Connell, The many roles of an RNA editor. Nat Rev
Genet, 2001. 2(11): p. 869-78.
48. Kume, H., et al., A-to-I editing in the miRNA seed region regulates target mRNA selection and silencing efficiency. Nucleic Acids Res, 2014. 42(15): p. 10050-60.
49. Seton-Rogers, S., MicroRNAs: Editing changes the meaning. Nat Rev Cancer, 2012. 12(12): p. 797.
50. Warnefors, M., et al., Conserved microRNA editing in mammalian evolution, development and disease. Genome Biol, 2014. 15(6): p. R83.
51. Bartel, D.P., MicroRNAs: genomics, biogenesis, mechanism, and function. Cell, 2004. 116(2): p. 281-97.
52. Iida, K., H. Jin, and J.K. Zhu, Bioinformatics analysis suggests base modifications of tRNAs and miRNAs in Arabidopsis thaliana. BMC Genomics, 2009. 10: p. 155.
53. Silberberg, G., et al., Deregulation of the A-to-I RNA editing mechanism in psychiatric disorders. Hum Mol Genet, 2012. 21(2): p. 311-21.
54. Sodhi, M.S., et al., RNA editing of the 5-HT(2C) receptor is reduced in schizophrenia. Mol Psychiatry, 2001. 6(4): p. 373-9.
55. Kubota-Sakashita, M., et al., A role of ADAR2 and RNA editing of glutamate receptors in mood disorders and schizophrenia. Mol Brain, 2014. 7: p. 5.
56. Pearce, B.D., Schizophrenia and viral infection during neurodevelopment: a focus on mechanisms. Mol Psychiatry, 2001. 6(6): p. 634-46.
57. Sham, P.C., C.J. MacLean, and K.S. Kendler, Risk of schizophrenia and age difference with older siblings. Evidence for a maternal viral infection hypothesis? Br J Psychiatry, 1993. 163: p. 627-33.
58. Di Costanzo, E. and F. Schifano, [Viral hypothesis of schizophrenia. Critical reflections]. G Clin Med, 1990. 71(5): p. 361-3, 366.
59. The dopamine hypothesis, viral theory of schizophrenia, and season of birth. Br J Psychiatry, 1988. 152: p. 429-32.
61. Crow, T.J., G.R. Taylor, and D.A. Tyrrell, Two syndromes in schizophrenia and the viral hypothesis. Prog Brain Res, 1986. 65: p. 17-27.
62. Shrikhande, S., et al., Cytomegalovirus and schizophrenia. A test of a viral hypothesis. Br J Psychiatry, 1985. 146: p. 503-6.
63. van Kammen, D.P. and L.E. DeLisi, The viral hypothesis of schizophrenia: smoke--but is there fire? Psychopharmacol Bull, 1984. 20(3): p. 523-5.
64. Libikova, H., J. Pogady, and S. Breier, [The viral hypothesis of schizophrenia. Results of virological research]. Cesk Psychiatr, 1983. 79(6): p. 361-75.
65. Torrey, E.F. and M.R. Peterson, The viral hypothesis of schizophrenia. Schizophr Bull, 1976. 2(1): p. 136-46.
66. Ota, H., et al., ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell, 2013. 153(3): p. 575-89.
67. Shoshan, E., et al., Reduced adenosine-to-inosine miR-455-5p editing promotes melanoma growth and metastasis. Nat Cell Biol, 2015. 17(3): p. 311-21.
68. George, C.X., M.V. Wagner, and C.E. Samuel, Expression of interferon-inducible RNA adenosine deaminase ADAR1 during pathogen infection and mouse embryo development involves tissue-selective promoter utilization and alternative splicing. J Biol Chem, 2005. 280(15): p. 15020-8.
69. Jabara, C.B., et al., Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc Natl Acad Sci U S A, 2011. 108(50): p. 20166-71.
70. Zhou, S., et al., Primer ID Validates Template Sampling Depth and Greatly Reduces the Error Rate of Next-Generation Sequencing of HIV-1 Genomic RNA Populations. J Virol, 2015. 89(16): p. 8540-55.
71. Eggington, J.M., T. Greene, and B.L. Bass, Predicting sites of ADAR editing in double-stranded RNA. Nat Commun, 2011. 2: p. 319.
72. Karolchik, D., A.S. Hinrichs, and W.J. Kent, The UCSC Genome Browser. Curr Protoc Bioinformatics, 2009. Chapter 1: p. Unit1 4.
73. Reuter, J.S. and D.H. Mathews, RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics, 2010. 11: p. 129.
74. Luciano, D.J., et al., RNA editing of a miRNA precursor. RNA, 2004. 10(8): p. 1174-7. 75. Wang, I.X., et al., ADAR regulates RNA editing, transcript stability, and gene expression.
Cell Rep, 2013. 5(3): p. 849-60.
76. Hwang, T., et al., Dynamic regulation of RNA editing in human brain development and disease. Nat Neurosci, 2016. 19(8): p. 1093-9.
TABLES AND FIGURES
Figure 1
The precursor to mature miR137 is predicted to undergo A-to-I RNA editing by ADAR1 at
significant levels. (A) This graph displays the output from InosinePredict, which represents
relative ADAR1-based editing likelihoods at the various positions along pre-miR137. The bars
colored in red represent sites that were previously hypothesized to be editing sites based on
in-house data and the purple base numbers represent implausible editing sites. (B) This figure
Table 1
Table 1a
Table 1b
MicroRNA 137 is subjected to A-to-I editing in vivo at multiple base positions. (A) This
table displays total editing rates by species at the three hypothesized edit sites in miR137, as
determined from high throughput RNA-sequencing data. (B) This table portrays the results of
further analysis of the RNA-sequencing data to determine frequencies of distinct edit
combinations. Strict combinations refer to edits solely at the enumerated sites (e.g. “1”
corresponds to editing at only site 1).
Index Table Raw Frequency Table
Index Position in pre-mir137 Base Edit Frequency in Adult Mouse Edit Frequency in Fetal Human
1 29 29 0.032675289 0.00046463
2 69 69 0.055820286 0.00092926
3 74 74 0.080326753 0.00139389
SPECIES
Strict Combination Table
Strict Indexed Edit Combination Actual Combination Frequency Predicted Combination Frequency Fold difference (Actual/Predicted)
1 0.024506467 0.028373157 0.863720137
2 0.039482641 0.049658991 0.795075381
3 0.061946903 0.073364702 0.844369306
12 0.002722941 0.001677432 1.623278902
13 0.004765146 0.002478188 1.922834589
23 0.012933969 0.004337351 2.981997362
123 0.000680735 0.000146511 4.646291942
SPECIES
Strict Combination Table
Strict Indexed Edit Combination Actual Combination Frequency Predicted Combination Frequency Fold difference (Actual/Predicted)
1 0.00046463 0.000463551 1.002327258
2 0.00092926 0.000927534 1.001861331
3 0.00139389 0.001391948 1.001395403
12 0 4.31E-07 0
13 0 6.47E-07 0
23 0 1.29E-06 0
123 0 6.02E-10 0
NOTE: Strict combination implies that non-specified indices are NOT edited NOTE: Strict combination implies that non-specified indices are NOT edited
ADULT MOUSE
Figure 2
A-to-I RNA editing is conserved at relatively high levels at two distinct positions present in
mature miR137. This graph illustrates the normalized editing rates (based on species-specific
averages) at all of the observed edit sites for each species. Positions 69 and 74 exhibit relatively
Figure 3
A-to-G modifications to miR137 impart changes in target affinity in vitro. This figure
displays the results of luciferase assays performed to quantify miR137 antisense target binding
affinity for different edit combinations. Luciferase activity is proportional to target gene
expression, so target affinity is inversely related to luciferase activity. The numeric combinations
and All correspond to various edit combinations, Control corresponds to a positive control (binds
strongly to the target), PL corresponds to a negative control (does not bind to the target), and WT
Figure 4
A molecular model of interactions that contribute to schizophrenia risk. This schematic
depicts the biochemical model constructed and subsequently simulated using MATLAB. The
curved lines represent RNA molecules, the brown circles represent A-to-I edits, standard arrows
Figure 5
The presence of the rs1625579risk SNP increases schizophrenia-related inflammatory
burden. This plot portrays the inflammatory burden output of the IFN-miR137-ADAR1
mathematical model with and without the rs1625579 risk SNP. The orange line corresponds to
the results in the presence of the risk SNP and the blue line corresponds to the results in the
Figure 6
Call:
lm(formula = IBFC ~ rEDIT, data = re)
Residuals:
Min 1Q Median 3Q Max -4.394e-05 -1.919e-05 -5.200e-08 1.923e-05 4.404e-05
Coefficients:
Estimate Std. Error t value Pr(>|t|) (Intercept) 1.045e+00 1.615e-05 64689 <2e-16 *** rEDIT 2.496e-02 1.664e-04 150 <2e-16 *** ---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
Residual standard error: 2.84e-05 on 12 degrees of freedom Multiple R-squared: 0.9995, Adjusted R-squared: 0.9994 F-statistic: 2.25e+04 on 1 and 12 DF, p-value: < 2.2e-16
A-to-I editing of miR137 significantly influences the effect of the rs1625579risk SNP on
schizophrenia-related inflammatory burden. This figure represents the output of the statistical
analysis on inflammatory burden fold change under different miR137 editing rate conditions.
The analysis was performed using a standard linear regression approach, where inflammatory
burden fold change was modeled using basal editing rate as the predictor (while all other factors
were held constant). The basal editing rate significantly affected inflammatory burden fold
Figure 7
A holistic schizophrenia risk model framework that incorporates genetic risk,
environmental risk, and gene-environment interaction. This figure represents a holistic
conceptual risk model framework for schizophrenia posited based on the findings of this study.
The enclosed terms represent causal or intermediary entities, while the arrows represent causal
APPENDIX
Supplementary Table 1
Multiple sites on miR137 are edited in vivo at levels significantly higher than background
changes. This table displays the base-modification read data from the high throughput
RNA-sequencing experiment that met significance criteria under a Poisson distribution. Reference
Base refers to the WT base and Error Base refers to the base read by the sequencer. Position Reference Base Consensus Count Tissue Error Base Error Base Count
29 A 8609 Fetal Human Brain G 4
41 A 8609 Fetal Human Brain G 4
43 A 8609 Fetal Human Brain G 3
44 A 8609 Fetal Human Brain G 4
50 A 8609 Fetal Human Brain G 4
53 A 8609 Fetal Human Brain G 5
61 A 8609 Fetal Human Brain G 4
69 A 8609 Fetal Human Brain G 8
71 A 8609 Fetal Human Brain G 4
72 A 8609 Fetal Human Brain G 12
74 A 8609 Fetal Human Brain G 12
29 A 1469 Adult Mouse Brain G 48
61 A 1469 Adult Mouse Brain G 14
68 A 1469 Adult Mouse Brain G 4
69 A 1469 Adult Mouse Brain G 82
71 A 1469 Adult Mouse Brain G 3
72 A 1469 Adult Mouse Brain G 11
Supplementary Figure 1
Response hsaACTB
Effect Tests
Source Nparm DF Sum of
Squares F Ratio Prob > F
Plasmid 9 9 37.535475 1.4211 0.2474
Least Squares Means Table
Level Least Sq
Mean
Std Error Mean
a119g 28.194555 0.9890734 28.1946
a119g-a124g 29.129404 0.9890734 29.1294
a124g 28.179814 1.2113626 28.1798
a79g 29.026718 0.9890734 29.0267
a79g-a119g 27.558155 0.9890734 27.5582
a79g-a119g-a124g 28.604111 0.9890734 28.6041
a79g-a124g 30.555158 0.9890734 30.5552
None 26.241238 0.9890734 26.2412
pCMVmiR137 27.264446 0.9890734 27.2644
pCMVPL 27.797063 0.9890734 27.7971
Response hsaGAPDH202
Effect Tests
Source Nparm DF Sum of
Squares
F Ratio Prob > F
Plasmid 9 9 50.368116 1.5206 0.2109
Least Squares Means Table
Level Least Sq
Mean
Std Error Mean
a119g 30.210362 1.1076074 30.2104
a119g-a124g 31.547063 1.1076074 31.5471
a124g 30.011102 1.3565365 30.0111
a79g 31.265220 1.1076074 31.2652
a79g-a119g 29.465520 1.1076074 29.4655
a79g-a119g-a124g 30.359163 1.1076074 30.3592
a79g-a124g 32.977412 1.1076074 32.9774
None 28.171171 1.1076074 28.1712
pCMVmiR137 29.198672 1.1076074 29.1987
pCMVPL 29.506629 1.1076074 29.5066
Response Pre-miR137
Effect Tests
Source Nparm DF Sum of
Squares
F Ratio Prob > F
Least Squares Means Table
Level Least Sq
Mean
Std Error Mean
a119g 22.618089 2.0149759 22.6181
a119g-a124g 25.152132 2.0149759 25.1521
a124g 22.239349 2.4678314 22.2393
a79g 22.484688 2.0149759 22.4847
a79g-a119g 22.994235 2.0149759 22.9942
a79g-a119g-a124g 22.360327 2.0149759 22.3603
a79g-a124g 23.163958 2.0149759 23.1640
None 28.677297 2.0149759 28.6773
pCMVmiR137 22.822852 2.0149759 22.8229
pCMVPL 28.764065 2.0149759 28.7641
The luciferase reporter-based target affinity data does not represent differences in
construct expression. This figure presents the output of a regression of the luciferase data
against RT-PCR data for each construct. All of the p-values exceed 0.05, which suggests that the
luciferase data is not explained by variation in expression of the various plasmids used in the
Supplementary Figure 2
The IFN-miR137-ADAR1 mathematical model exhibits biologically plausible input-output
dynamics across time. This graph displays the results of a simulation of the
IFN-miR137-ADAR1 mathematical model for a periodic infection input. Pathogen levels (corresponding to
the periodic infection input) are in blue.
Supplementary Computational Methods
The square-wave pathogen input function was constructed in MATLAB as follows:
function [out] = SquareWave (t, period, length, high, low)
if (mod(t,period) <= length)
out = high; else
out = low; end
The ODE system employed in this study was implemented in MATLAB as follows:
function [dydt] = IFNmir137Model(t,y,parameters)
% Order of Quantities: Pathogen, IFN, ADAR1-A Level, Total miR137, TCF4 % mRNA, TCF4 protein, Dendritic Cells, Inflammation Burden.
% IFN degradation rate, ADAR1-A synthesis rate per IFN, ADAR1-A degradation rate,
% basal miR137 transcription rate, binary rs1625579 % presence, miR137 degradation rate, basal
% editing rate, ADAR1-B level, TCF4 transcription rate,
% TCF4 degradation rate, miR137-TCF4 targeting Vmax, miR137-TCF4 targeting % Km, TCF4 protein production Vmax, TCF4 protein production Km, TCF4
% protein degradation Vmax, TCF4 protein degradation Km, DC basal production rate, DC replication rate per TCF4 protein,
% DC degradation rate, Inflammation factor.
dydt = zeros(8,1); dydt(1) =
SquareWave(t,parameters(1),parameters(2),parameters(3),parameters(4)) - parameters(5)*y(1);
dydt(2) = parameters(6)*y(1)*y(7) - parameters(7)*y(2); dydt(3) = parameters(8)*y(2) - parameters(9)*y(3);
dydt(4) = parameters(10)*(1 - 0.3*parameters(11))*(1 + 2.98*(y(3)/20)) - parameters(12)*y(4);
E = parameters(13)*(1 + (0.6*y(4))/parameters(14)); dydt(5) = parameters(15) - parameters(16)*y(5) -
.16*(E*y(4)*y(5)*parameters(17))/(parameters(18)+y(5)) - ((1-E)*y(4)*y(5)*parameters(17))/(parameters(18)+y(5));
dydt(6) = (parameters(19)*y(5))/(parameters(20) + y(5)) - (parameters(21)*y(6))/(parameters(22)+y(6));
dydt(7) = parameters(23) + parameters(24)*y(6) - parameters(25)*y(7); dydt(8) = parameters(26)*y(2);
end
The sensitivity analysis method was implemented in MATLAB as follows:
function [out] = SNPcompare
(t,y,tspan,y0,want,test,parameters,varySet,lower,upper,steps)
% Initialize inputs and outputs
set = varySet;
curr = parameters(set);
currL = curr*(1-(lower/100)); currU = curr*(1+(upper/100));
for idx = 1:length(curr)
vec = linspace(currL(idx),currU(idx),steps);
if idx == 1
mat = vec;
else
mat = combvec(mat,vec); end
end
mat = mat';
out = [mat, zeros(size(mat,1),1)]; progressbar;
% Scan through parameter values and compute the output metrics
for idx = 1:size(mat,1)
parms = parameters;
yS0 = yn(end,want); parms(test) = 1;
[~,yn] = ode45(@(t,y) IFNmir137Model(t,y,parms),tspan,y0); yS1 = yn(end,want);
out(idx,end) = yS1/yS0;
progressbar(idx/size(mat,1)); end