VERSATILE ROUTES TO PHOTO-RESPONSIVE POLYESTERS FOR DUAL AND TRIPLE SHAPE MEMORY BIOMATERIALS
Jason Michael Rochette
A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the
Department of Chemistry.
Chapel Hill 2012
Approved by
Dr. Valerie Sheares Ashby
Dr. Wei You
Dr. James Cahoon
Dr. Joseph Templeton
ii
© 2012
iii
ABSTRACT
JASON M. ROCHETTE: VERSATILE ROUTES TO PHOTO-RESPONSIVE POLYESTERS FOR DUAL AND TRIPLE SHAPE MEMORY BIOMATERIALS
(Under the direction of Dr. Valerie S. Ashby)
Shape memory polymers are promising materials for smart biomedical devices and applications, however, thermal systems are limited by a narrow applicable temperature range. Light-induced shape memory (LSM) circumvents this limitation by using a mechanism
iv
This work is dedicated to my family, Susan Rochette, Neil Rochette, Dr. Jennifer
Rochette, and David Rochette for their love, support, and motivation, as well as my
colleagues and friends, Dr. Hayden Black, Dr. Duy Le, Sarah Brosnan, Annie Jackson, Dr.
Peter Uthe, Dr. Benjamin Pierce, Dr. Tim Merkel, Dr. Bryan Frauhiger, and Stuart Dunn for
their advice, intellectual discussions, and friendships. I would like to thank my advisor, Dr.
Valerie S. Ashby for her direction, encouragement, and trust. I would also like to thank my
friends, Landon Simmons, Mike Vysocka, Mike Hargrove, Matt Rivenbark, Antoine Moore,
and Michael Downer for their lasting friendships, support, and diversions from work needed
periodically. Most importantly I would like to thank and dedicate this work to Amber Baxley
for standing by me throughout my graduate career amidst the long nights and weekends in
lab, the grumpy moods when experiments weren’t working, acting as my one-woman
audience as I prepared for presentations and interviews, and for her endless love and
v
TABLE OF CONTENTS
LIST OF TABLES ...x
LIST OF FIGURES ... xi
LIST OF ABBREVIATIONS ... xiii
LIST OF SYMBOLS ... xvii
Chapter I LITERATURE REVIEW OF SHAPE MEMORY MATERIALS ...1
1.1 The Shape Memory Effect ...1
1.2 Shape Memory Materials ...3
1.2.1 Thermoplastic Shape Memory Polymers ...3
1.2.2 Thermoset Shape Memory Polymers ...5
1.2.3 Composites and Liquid Crystalline Shape Memory Polymers ...7
1.2.4 Shape Memory Biomaterials...8
1.3 External Stimuli ...9
1.3.1 Thermal Triggering ...9
1.3.2 Indirect Thermal Actuation ...10
1.4 Light-Responsive Polymers ...13
1.4.1 Photochromic Molecules and Photo-isomerization ...14
1.4.2 Light-Induced Shape Changing ...15
1.4.3 Light-Induced Shape Memory ...16
vi
1.6 Conclusions ...21
1.7 Dissertation Organization ...21
References ...23
II PHOTO-RESPONSVIE POLY(ESTER URETHANE)S FOR SHAPE MEMORY BIOMATERIALS ...31
2.1 Introduction ...31
2.2 Experimental Section ...34
2.2.1 Materials ...34
2.2.2 Instrumentation ...34
2.2.3 Monomer Synthesis ...35
2.2.4 Polyester Prepolymer Synthesis ...36
2.2.5 Thermoset Network Formation ...39
2.2.6 Characterization ...40
2.2.6.1 Light-induced Shape Memory (LSM) ...40
2.2.6.2 Physiological Degradation and Water Uptake ...40
2.2.6.3 Cytotoxicity Studies ...41
2.3 Results and Discussion ...42
2.3.1 Monomer Synthesis ...42
2.3.2 Polymer Synthesis ...45
2.3.3 Thermal and Mechanical Analysis ...46
2.3.4 Degradation and Cytotoxicity ...49
2.3.5 Light-induced Shape Memory ...52
2.4 General Conclusions ...55
vii
References ...57
III MULTIFUNCTIONAL TRIPLE SHAPE MATERIALS COMBINING LIGHT AND THERMAL TRIGGERS ...58
3.1 Introduction ...58
3.2 Experimental Section ...60
3.2.1 Materials ...60
3.2.2 Instrumentation ...60
3.2.3 Light-induced Shape Memory Characterization ...61
3.2.4 Thermal Shape Memory Characterization ...61
3.2.4.1 Macroscale Shape Memory...61
3.2.4.2 Microscale Surface Shape Memory ...61
3.3 Results and Discussion ...62
3.3.1 Thermal Shape Memory ...62
3.3.1.1 Delocalized Macroscopic Shape Memory ...62
3.3.1.2 Localized Surface Shape Memory ...64
3.3.2 Triple Shape Memory ...66
3.3.2.1 Combination Macroscopic/Macroscopic Triple Shape Memory ...68
3.3.2.2 Combination Macroscopic/Microscopic Triple Shape Memory ...69
3.4 General Conclusions ...70
3.5 Acknowledgements ...71
References ...72
viii
4.1 Introduction ...73
4.2 Experimental Section ...75
4.2.1 Materials ...75
4.2.2 Instrumentation ...75
4.2.3 Functional Monomer Synthesis ...76
4.2.3.1 Functional Amine Monomer ...76
4.2.3.2 Functional Diacrylate Monomer ...77
4.2.4 Polymer Synthesis ...78
4.2.4.1 Post-Polymerization Route ...79
4.2.4.2 Functional Monomer Route ...82
4.2.5 Thermoset Network Formation ...83
4.3 Results and Discussion ...84
4.3.1 Functional Monomer Synthesis ...84
4.3.1.1 Amine Monomer ...84
4.3.1.2 Diacrylate Monomer ...85
4.3.2 Polymer Synthesis ...86
4.3.2.1 Post-Polymerization Route ...87
4.3.2.2 Functional Diacrylate Route ...90
4.3.3 Thermal and Mechanical Analysis ...92
4.3.3.1 Post-Polymerization Route ...92
4.3.3.2 Functional Diacrylate Route ...94
4.3.3.3 PBAE Elastomers...95
ix
4.5 Acknowledgements ...97
References ...98
V GENERAL CONCLUSIONS AND FUTURE RESEARCH DIRECTIONS ...99
5.1 General Conclusions ...99
5.2 Directions ...100
References ...101
Appendix A: SUPPLEMENTAL MATERIALS FOR CHAPTER 2 ...103
Appendix B: SUPPLEMENTAL MATERIALS FOR CHAPTER 3 ...146
Appendix C: SUPPLEMENTAL MATERIALS FOR CHAPTER 4 ...151
x
LIST OF TABLES
Table
1.1 Effect of crosslinking on mechanical and shape memory
properties of crosslinked PUs ...7 2.1 Molecular weight and thermal characterization of photo-
responsive prepolymers ...47 2.2 Thermal and mechanical properties of photo-responsive
thermoset networks ...48 2.3 Characterization of light-induced shape memory in
DCA-OD and DCE-CHDM series ...54 3.1 Thermal shape memory characterization ...63 4.1 Molecular weight and thermal characterization of PBAE
prepolmers...93 4.2 Molecular weight and thermal characterization of CAD-OH
and CAD-Alkyne prepolymers ...95 4.3 Thermal and mechanical properties of PBAE thermoset
xi
LIST OF FIGURES
Figure
1.1 Programming and recovery of a general shape memory
polymer ...1
1.2 Strain fixity ratio (Rf) and strain recovery ratio (Rr) ...2
1.3 Components of segmented PCL based PEU SMP ...4
1.4 Amorphous thermoset copolyester urethane SMP ...6
1.5 SMP composite made by electrospun PCL fibers in Sylgard 184 matrix (top). Recovery of programmed temporary shape over 8 seconds at 80 oC (bottom) ...8
1.6 Electro-active shape memory polymer recovering from temporary unwound form to curled shape when voltage applied ...11
1.7 Shape memory PU immersed in water to trigger recovery by plasticization effect of water molecules ...12
1.8 Directed bending by linear polarized light irradiation of azobenzene LCEs at different angles ...15
1.9 Light-induced shape memory mechanism for crosslinked network possessing photoreactive cinnamate groups ...18
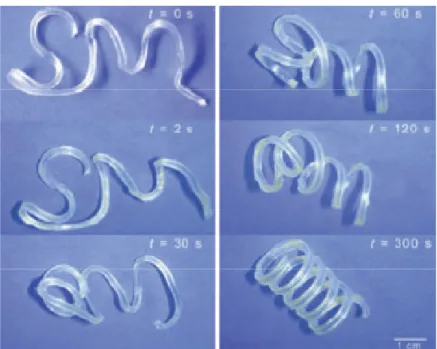
1.10 Recovery of triple shape polymer network by heating to 40 oC and 60 oC sequentially ...20
2.1 Light-induced shape memory mechanism ...33
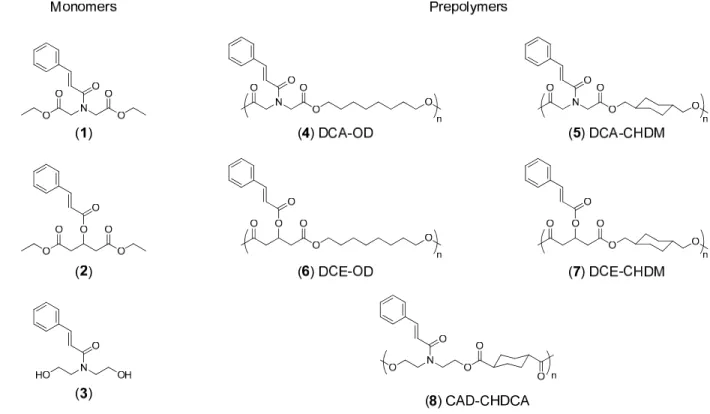
2.2 Bifunctional monomers DCA, DCE, CAD and resulting prepolymers from polycondensation with complementary monomers ...38
xii
2.4 Physiological hydrolytic degradation of DCA-OD and
DCE-CHDM in PBS (pH = 7.4) at 37 oC ...49 2.5 Water uptake for DCA-OD and DCE-CHDM ...50 2.6 Cell viability measured by CellTiter-Glo® ATP luminescence
assay. Relative luminescence count (top), percent of control
count (bottom) ...51 2.7 Macroscopic light-induced shape memory of DCA-OD
network ...53 3.1 Triple shape memory cycle ...58 3.2 Macroscopic thermal shape memory of DCA-OD (top) and
DCA-CHDM (bottom) networks ...64 3.3 Bright-field microscope images of microscopic thermal
shape memory of DCA-OD (top) and DCA-CHDM
(bottom) networks, scale bar = 10 µm ...65 3.4 Effect of photo-fixing on glass transition temperature of a
thermoset DCE-CHDM film ...67 3.5 Macroscopic triple shape programming/recovery process
combining light-induced shape memory and thermal shape memory (illustration, left). DCA-OD system following
programming and recovery process (images, right) ...68 3.6 Triple shape programming/recovery process combining
macroscopic and microscopic shape changes (illustration, left). DCA-OD system following programming and recovery process (images, right). Expansion: bright-field
microscopic surface images, scale bar = 10 µm ...70 4.1 Functional monomers ...78 4.2 Hydoxy pendant prepolymers and cinnamate (CA)
functionalized prepolymers ...81 4.3 GPC trace of EO-HD (blue) and EC-HD (orange) ...89 4.4 1H-NMR spectra of the methylene region of EO-HD (top)
xiii
LIST OF ABBREVIATIONS
AA Adipic Acid
AIBN 2,2’-Azobis(2-methylpropionitrile)
BC Butyl cinnamate
BD 1,4-Butanediol diacrylate BDO 1,4-Butanediol
BO Butanolamine CA Cinnamate
CAD N,N-bis(2-hydroxyethyl)cinnamamide CHDCA 1,4-Cyclohexane dicarboxylic acid
CHDM 1,4-Cylcohexanedimethanol
CNT Carbon nanotube
DA Diacrylate
DBU 1,8-Diazabicylo[5.4.0]undec-7-ene DCA Diethyl 2,2’-(cinnamoylazanediyl) diacetate DCE Diethyl 3-(cinnamoyloxy)pentanedioate DCM Dichloromethane
DI Deionized
DMA Dynamic mechanical analysis DMF Dimethylformamide
DP Degree of polymerization
xiv
EC Ethyl cinnamate
EO Ethanolamine Et2O Diethyl ether Et3N Triethylamine EtOAc Ethyl acetate
Fmoc 9-Fluorenylmethoxycarbonyl GPC Gel permeation chromatography HD 1,6-hexanediol diacrylate IR Infrared
LC Liquid crystalline LCE Liquid crystalline elastomer LSM Light-induced shape memory
MA Methyl acrylate
MDI 4,4’-diphenylmethane diisocyanate MeOH Methanol
ML Mass loss
MMA Methyl methacrylate MSA Mercaptosuccinic acid
MW Molecular weight
xv
PBAE Poly(β−amino ester) PBS Phosphate buffered saline PCHMA Poly(cyclohexyl methacrylate) PCL Polycaprolactone
PDI Polydispersity index PE Polyethylene PEG Poly(ethylene glycol) PEO Poly(ethylene oxide) PET Polyethylene terephthalate PEU Poly(ester urethane) PFPE Perfluoropolyether PGA Polyglycolide PLA Poly(lactic acid) PLLA Poly(L-lactide)
PMMA Poly(methyl methacrylate)
P-PEU Photo-responsive poly(ester urethane)s PRINT Particle replication in non-wetting templates PS Polystyrene
xvi
Sc(OTf)3 Scandium trifluoromethanesulfonate SCF Short carbon fiber
SCP Shape changing polymer SMA Shape memory alloy SME Shape memory effect SMP Shape memory polymer SnOct Tin (II) ethylhexanoate SSM Surface shape memory
TGA Thermogravimetric analysis THF Tetrahydrofuran
TPE Thiol pendant copolyesters TSM Triple shape memory TSP Triple shape polymer UV Ultraviolet
xvii
LIST OF SYMBOLS
Rf Strain fixity
Rr Strain recovery
εu Temporary relaxed strain
εm Maximum deformation strain
εp Residual strain after recovery
εmax Maximum deformation Ttrans Transition temperature
Tm Melting temperature
Tg Glass transition temperature
Th Higher temperature
Tl Lower temperature
Mn Number average molecular weight Mw Weight average molecular weight
G Young’s modulus
δ NMR shift
λ Wavelength
1.1 T appli with equip progr Reco mater shape switc Figur LIT
The Shape M
Shape me cation of an a wide range pment9,10,11, a rammed into
very occurs rial to the or e memory ef ches (switchi
re 1.1 Progr
TERATURE
Memory Eff
emory behav external stim e of applicat and textiles1 o a predeterm
in response riginal shape ffect (SME), ing segments ramming and E REVIEW fect
vior is define mulus.1 As s tions includi
2,13. The ma mined tempo
to the extern e (Figure 1.1
the system s or groups)
d recovery of
Chapter I OF SHAPE
ed as the cap such shape m ng biomedic aterial in its orary shape b
nal stimulus ). In order f is required t .14 Netpoint
f a general s
MEMORY
pability to ch memory mat cal devices2,3
permanent f by a stress or or switch th for a polyme to contain ne ts, whether p
shape memor
MATERIAL
hange shapes terials are sm
3,4,5, sensors6 form is packa r force and f hat actuates r eric material etpoints and physical or c
ry polymer. LS
s upon mart material
6,7,8, aerospa aged or fixed in this s
2
crosslinks, determine the permanent shape and allow for the memory of the original form. Between these netpoints are chain segments which act as molecular switches in response to specific stimuli that reversibly fix the temporary shape. Reversible fixing is possible through vitrification, crystallization, or formation of temporary covalent bonds.
The polymer segments must also have the ability to assume a variety of
conformations to allow deformation of the system in the programming process. In the fixed temporary shape the access to different conformations in the switching segments is limited creating a highly unfavorable entropic state. When the stimulus is applied, the fixation of the chain segments is reversed and reconfiguration to the maximum state of entropy drives the recovery of the original shape.15 The SME is quantified by the strain fixity ratio (Rf) and strain recovery ratio (Rr) (Figure 1.2), which evaluates the fixation of the temporary shape and the recovery of the original shape, respectively. Rf measures the ratio of strain in the relaxed temporary state (after unloading) (εu) to the maximum deformation (before
unloading) (εm) of the applied stress in programming. Rr measures the ratio of strain released during the recovery process (εu-εp) compared to the strain held in the packaged state (εu).16
Shape memory polymers (SMPs) should not be confused with another class of stimuli-responsive materials known as shape changing polymers (SCPs). SCPs differ from SMPs in two ways; first the temporary shape change in SCPs is a response to a stimulus
100 100
3
whereas the temporary shape in SMPs must be programmed by an external force or stress. Secondly, in SCPs exposure to the stimulus is required to keep the temporary shape change; once the stimulus is removed, the system returns to the original form.14 In SMPs the
programmed temporary shape is persistent until reapplication of the stimulus triggers the controlled recovery of the original shape.
1.2 Shape Memory Materials
The shape memory effect was first observed in metal alloys in the early 1950s with the study of plastic deformation and diffusionless phase changes in gold-cadmium alloys in response to a thermal stimulus.17 Other alloys such as Ni-Ti, Cu-Zn-Al, and Fe-Mn-Si have found technical application as shape memory materials for biomedical devices, electrical devices, and construction materials, respectively. While some possess the SME, shape memory alloys (SMAs) lack an elastic nature which limits the deformations (εmax = 8%) and shapes available for a given application.
The extension of the SME into polymeric materials such as thermoplastic or
thermoset elastomers has not only expanded the accessible shapes/deformations, but has also led to the development of diverse chemical and physical material property combinations, various external triggers, and expansive applications.18 The first example of a polymer system possessing the SME was crosslinked polyethylene (PE) via ionizing radiation, which came to be known as “heat-shrinkable tubing” in the 1960s.19,20 Since that time different polymers, copolymers, architectures, and composites have been utilized.
1.2.1 Thermoplastic Shape Memory Polymers
4
designated by the nature of the netpoints within the system which are physical crosslinks. PU SMPs are a two phase system consisting of hard and soft segments. In order to possess the SME, the hard segments must aggregate to form physical crosslinks which hold the permanent shape, while the soft segments act as switching segments. Hard segments are usually created from diisocyanates and chain extenders or from a macro-diol with a higher thermal transition.26 Soft segments can be low molecular weight diols, hydroxyl terminated oligomeric esters, or hydroxyl terminated oligomeric ethers. An example of such a
combination is a polyesterurethane (PEU) where oligourethane units act as the hard segment and polyesters as the soft segment. Kim and coworkers synthesized a series of
polycaprolactone (PCL) based PEUs with 4,4’-diphenylmethane diisocyanate (MDI) and 1,4-butanediol (BDO) to study the influence of the length and content of the PCL macrodiol on
5
shape memory properties (Figure 1.3).27 They showed there was a lower limit of PCL
molecular weight (MW) for crystallization to occur when in the segmented PU, which in turn allowed for the SME of the system.
Other linear block copolymers have also been studied as thermoplastic elastomers such as styrene-trans-butadiene-styrene triblocks28, poly(norbornene)-b -poly(norbornenyl-polyhedral oligosilsesquioxane)29, and polyethylene terephthalate-b-polyethylene oxide (PET-PEO)30. Miscible polymer blends such as poly(lactic acid)/poly(vinyl acetate)
(PLA/PVAc) and poly(methyl methacrylate)/poly(vinylidene fluoride) (PMMA/PVDF) also possess the SME due to one polymer imparting a hard phase and the other a soft phase.31 Ultra high MW polymers can also function as physically crosslinked materials when the entanglements act to hold the permanent shape.32 Although block copolymers, blends, and high MW polymers demonstrate the shape memory effect, the thermoplastic elastomer PU SMPs have the highest shape recoverability, a wider range of transition temperatures, better processing ability, and better biocompatibility depending on composition.33,34 One limitation of thermoplastic SMPs is their susceptibility to irreversible deformation and creep during the programming process.
1.2.2 Thermoset Shape Memory Polymers
6
Figure 1.4 Amorphous thermoset copolyester urethane SMP.
properties.35 The covalent network of thermoset elastomers can be achieved by crosslinking of linear or branched prepolymers, or polymerization in the presence of at least one
trifunctional or greater monomer.36-38 As mentioned earlier, the first example of this type of system was crosslinked PE, but other homopolymers, copolymers, and blends also show the SME when crosslinked such as PE/poly(vinyl alcohol) (PVA)36, poly(vinyl chloride)
(PVC)38, PCL39, PS40, and PEO-PET41. Thermoplastic PU and PEU SMPs (Figure 1.4) have also been improved by introducing covalent crosslinks via incorporation of a trifunctional diol as a chain extender or by end-functionalizing a precursor with crosslinkable end
7
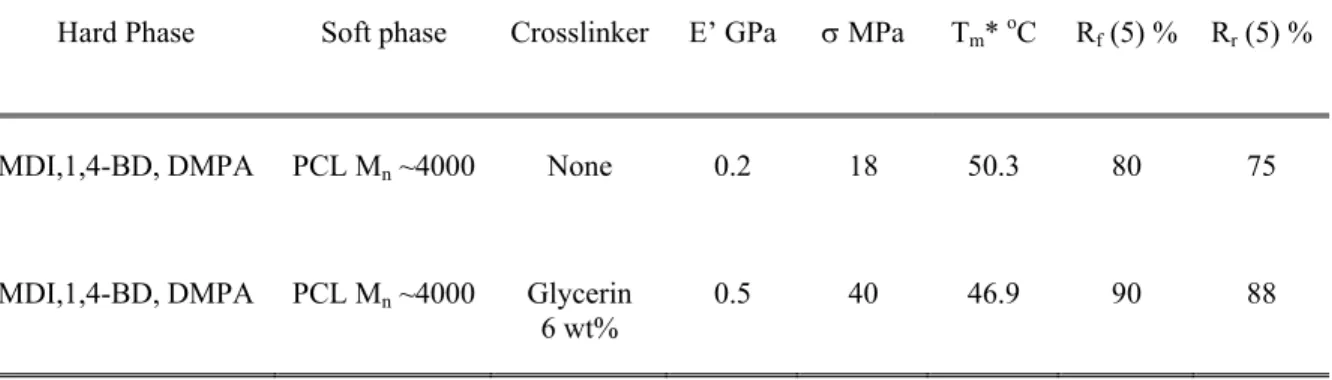
Table 1.1 Effect of crosslinking on mechanical and shape memory properties of crosslinked PUs.45,42
Hard Phase Soft phase Crosslinker E’ GPa σ MPa Tm* oC Rf (5) % Rr (5) %
MDI,1,4-BD, DMPA PCL Mn ~4000 None 0.2 18 50.3 80 75
MDI,1,4-BD, DMPA PCL Mn ~4000 Glycerin
6 wt% 0.5 40 46.9 90 88
*melting transition of soft phase
1.2.3 Composites and Liquid Crystalline Shape Memory Polymers
Besides thermoplastic and thermoset polymers/copolymers, there has also been the development of liquid crystalline SMPs and composite SMPs to diversify the possible applications. Incorporation of fillers into SMPs is utilized to reinforce material strength, improve mechanical properties, create conductive composites, and diversify triggering methods.35,46,47 Fillers in these composites include fibers48-50, glass51, carbon black52, silicon carbide53,5, carbon nanotubes (CNTs)54-63, or metallic particles64-68. Distinct fabrication methods can also be employed to produce SMP composites made from only polymer
material. Mather et al. used electrospinning to create a nonwoven fiber mat of PCL within a silicon rubber matrix16 (Figure 1.5).
Figur Reco 1.2.4 has b PCL-temp SMA that c comb polye hydro have therm norm
re 1.5 SMP very of prog
Shape Mem
SMPs spa been an area
-based biode eratures by c A had found a could be com binations allo ethylene glyc ophilicity, pe been used fo moplastic and mally more op
P composite m grammed tem
mory Bioma
an a wide ran of considera egradable the controlling t application f mprised of PC
owed for deg col (PEG) to ermeability, or biodegrad d thermoset paque. Fully
made by elec mporary shap
aterials
nge of poten able research ermoplastic the PCL mol for self-depl CL, polygly gradable dev o the compos and degrada dable SMPs, SMPs using y amorphous 8 ctrospun PC pe over 8 sec
ntial applicat h interest.74-7 SMP that co lecular weigh
oying stents colide (PGA vices that req sition improv ability.80,81 C depending o g a Tm switch
s SMPs, on t
CL fibers in S conds at 80 o
ions, howev 76 This bega ould be targe ht and hard/ , but the intr A), poly(L-la quired no rem
ved properti Crystalline a on the target h are semi-cr the other han
Sylgard 184 oC (photos, b
ver biomedic an with the d eted for spec soft phase co roduction of actide) (PLLA
moval.78,79 T ies such as in and amorpho
t application rystalline an nd, are trans
matrix (top) bottom).16 cal applicatio development ific switchin ompositions f synthetic SM
9
meet the requirements for ocular tissue transplants.82,83 Degradation rates can also be tailored depending on the monomers incorporated. PCL materials alone have a low biodegradation rate, but as mentioned previously incorporation of PEG can increase the rate. PLA based materials also show faster degradation, however, the thermal transition cannot be targeted to temperatures close to body temperature (37 oC), which is necessary for SMP activation in biomedical devices.84
1.3 External Stimuli
To this point nearly all examples of SMPs discussed possess thermal shape memory that is triggered by heating, however there are a variety of other triggers or external stimuli that can be used to switch a packaged shape to the original shape including magnetic, electric, solvent, and light stimuli.
1.3.1 Thermal Triggering
The most common and extensively studied SMP mechanism is based on thermal transitions where a material is heated above a transition temperature (Ttrans) actuating a shape change from temporary to permanent. The specific mechanism for thermal shape memory is dependent on the type of material (thermoplastic or thermoset) and the thermal transition (melting transition, Tm, or glass transition, Tg).
10
based thermoplastic SMP, the system is heated above Ttrans (< Th) and deformed by an external force. In this deformed state, the soft segment polymer chains have been
reassembled into an entropically less favorable conformation and upon cooling below Ttrans the soft segments recrystallize, confining the polymer chains in this entropically strained position and fixing the temporary shape. When heated above Ttrans again, the newly arranged crystallites of the soft phases melt and the physical netpoints of the hard segment recover to the original position.
For thermoplastic SMP systems based on Tg switches, the above mechanism is followed, instead of the soft segments crystallizing to hold the entropic strain, they vitrify which can lead to a small decrease in strain fixity. The recovery rate can also be slower for Tg based materials due to the broad thermal transition compared to the sharp melting transition of a semicrystalline soft segment.
Thermoset SMPs on the other hand do not require multiple phases in order to possess a thermal SME, however multiphase compositions like thermoset PEUs are still applicable. In either case chemical crosslinks hold the permanent network so Ttrans can be the highest thermal transition in the system without losing the original shape. Systems based on a Tm or Tg behave in the same manner as thermoplastic SMPs with either reversible crystallization or vitrification to fix the temporary shape after physical deformation while heated above Ttrans followed by cooling.
1.3.2 Indirect Thermal Actuation
11
fields85,86, electrical current87,88, infrared (IR) irradiation3,89, and water/solvent interactions 90-93 are able to trigger a thermal mechanism in specifically designed SMP materials and composites. As mentioned earlier, composites can contain an assortment of fillers, and it is this availability of various fillers in SMP composites that accounts for the variety of stimuli which can be utilized.
Electro-responsive SMP composites can be filled with conductive materials such as carbon nanotubes94-96, carbon particles (carbon black)97,52, Ni powder98, and short carbon fibers (SCF)99-100. When a voltage is applied the conducting materials will produce resistive heating and the surrounding SMP material will be thermally actuated.35 Goo et al. reinforced a shape memory PU system with multi-walled carbon nanotubes (MWNTs) and showed electro-active shape recovery with 10.4 % energy conversion efficiency, as well as recovery rates as fast as 10 seconds for macroscopic shape changes when a constant voltage was applied (Figure 1.6).101 Magnetic-responsive composites demonstrate similar recovery when placed within an alternating magnetic field, triggering inductive heating.102 The composite fillers in these cases are magnetic iron oxide particles (Fe2O3 or Fe3O4)
12
Figure 1.7 Shape memory PU immersed in water to trigger recovery by plasticization effect of water molecules.91
homogenously distributed throughout the polymer matrix.85,103 Shape memory composites impregnated with tuned gold nanoparticles can also be actuated via IR laser irradiation at specific wavelengths.104
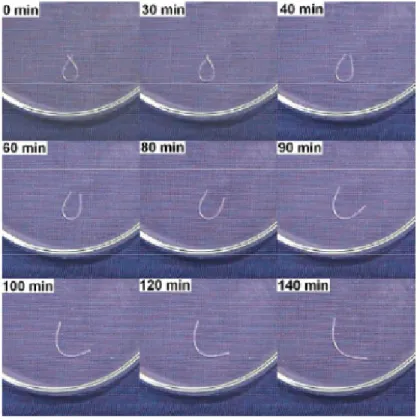
Other methods of indirect thermal actuation can be achieved in non-composite materials, however in some cases the addition of fillers will enhance the response rate. IR irradiation can be used as a source of radiant thermal energy when a SMP is illuminated with an IR laser or optical fiber105, and the addition of carbon black to SMPs increases the
13
When the effect is pronounced enough to lower Ttrans below the ambient temperature as in the case of PU in water91 (Fiugre 1.7) or styrene –based SMPs in DMF109 or toluene, recovery begins to occur albeit at a slow rate depending on the rate of solution penetration in the material.
Recently, polymer networks containing metal-ligand coordination complexes have been studied as responsive metallo-supramolecular materials, which can be triggered with heat, light, or chemical triggers.110 The materials are based on a crosslinked poly(butadiene) network endcapped with ligands, which when in the presence of metal salts will form
supramolecular reversible crosslinks. Heating of the metal-ligand coordination bonds decomplexes the metal center and cleaves the reversible crosslinks. The combination of triggers that can activate this system are related to different methods mentioned previously: normal heating, UV light absorption leading to localized heating, and chemical/solvent interactions interfering with the thermal stability of the material via plasticization or decomplexation.
1.4 Light-Responsive Polymers
14
Scheme 1.1 Photochromic molecules: azobenzene (1) spirobenzopyran (2) triphenylmethane leuconitrile (3) cinnamic acid (4).
1.4.1 Photochromic Molecules and Photo-isomerization
15
photoisomerizations include azobenzene(1)118-120, stilbene121, triphenylmethane
leucoderivatives(2)116, spirobenzopyrans (3)122, hydroxytriphenylmethanol117, coumarin, and cinnamic acid (4).
1.4.2 Light-Induced Shape Changing
Extension of the molecular changes induced by photoisomerization to a macroscopic level leads to light-induced shape changes in polymers and gels such as bending123-125, contraction126,127, and volume change128-131 in isolated materials under no force of
16
deformation when under light irradiation. When the light source is removed, the shape change generally reverses.123-127,132-134 Polymer gels with triphenylmethane leucoderivatives incorporated swelled in the presence of UV light due to the formation of cyanide ions
creating an osmotic pressure change, and when the light was removed, the gel shrank.129 The dissociation into ions also creates an electrostatic repulsion effect. Photo-responsive
elastomers containing azobenzene in the polymer backbone or side chains can reversibly contract and expanded based on the light-induced cis-trans isomerization under two different UV wavelengths.112,135,136 The development of azobenzene LCEs has led to larger
macroscopic changes such as bending due to the ordered nature of the materials125,133,137,138, which also allows for direction-controlled bending123 using linear polarized light at different angles (Figure 1.8).
1.4.3 Light-Induced Shape Memory
The controlled bending of azobenzene LCEs in one of the few examples of somewhat of a predetermined shape change for light-induced shape changing polymers, however as mentioned previously, the removal of the light source recovers the original form. In order to allow programming of a predetermined temporary shape and triggered recovery, a
mechanism to fix the material based on a photoisomerization is necessary. Recently, White
et al. demonstrated a light-induced shape memory effect in azobenzene LCEs by programming with circular polarized light.139 The angle of polarized light allows for
17
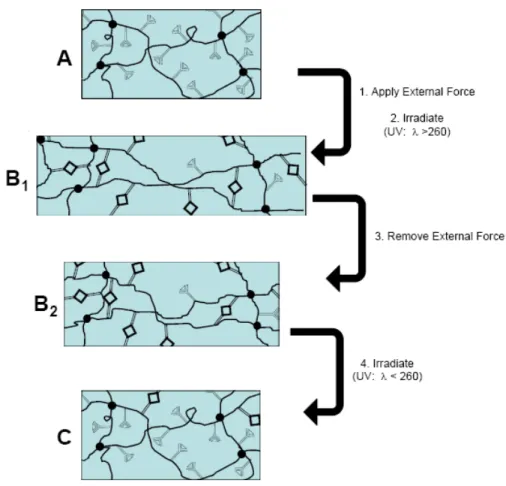
Scheme 1.2 Light-induced SMP synthesized by Lendlein et al.
limitation on the temporary programming available remains because temporary shapes are restricted to bending at angles between -25o – 25o. The first light-induced SMPs were developed by Lendlein et al., which could be physically deformed and fixed into a variety of shapes (comparative to thermal SMPs), based on photoreactive cinnamate groups which undergo reversible photo-induced [2+2] cycloaddition.140 The material was a thermoset hydrogel synthesized by the radical-initiated polymerization of various acrylate and
Figur photo netpo locke relax up to cross induc remo
re 1.9 Light oreactive cin
oints will rel ed out from r
ation leads t 60 min. Str links are cle
As is the c ced SMPs is
tely at ambi
-induced sha nnamate grou
ax to the poi relaxation by to strain fixit rain recovery eaved during
case with all biomedical ent temperat
ape memory ups.
int at which y crystallizat ty values of y is compara g irradiation w
l types of SM devices, giv tures.140 Rec
18 y mechanism
they are hel tion or vitrif 20-50% in l able to therm
with the high MPs, a major ven that light cently, Wu e
m for crosslin
d, whereas s fication in th ight-induced mal SMPs, al her energy w r application t-induced tri
et al. synthes
nked network
switching se hermal SMPs
d SMP syste lthough not a wavelength o n field of inte ggering can sized a therm
k possessing
gments are s. This
ms irradiate all temporar of light. erest for ligh
19
Scheme 1.3 Light-induced thermoplastic PEU SMP synthesized by Wu et al.
1.5 T more when trigge the fi the sa switc differ withi differ meth a PCL respe Figur seque Triple Shape The SME complex m n programme ering recove irst temporar ame network ches can be a rent phases14 in a broad tra The first t rent polymer acrylate) (PC L network w ectively. Fig
re 1.10 Reco entially.
e Memory
E can be exte ovement. T ed in a multi ery resulting ry shape.142 k, each contr a combinatio 44-146, or it ha ansition can triple shape p r architectur CHMA) seg with PEG sid gure 1.10 sho
overy of trip
ended from d SM systems i-step proces in the origin Most TSM s ributing an in on of two me
as also been also result in polymers (T es.143 The fir ments as a r de chains (CL
ows an exam
ple shape pol
20 dual to triple s possess two ss the materi
nal shape fro systems poss ndependent elting and/or shown that n independe TSPs) were d rst contained andom copo LEG) giving mple of the C
lymer netwo
shape mem o independen
al can hold t om the secon sess two pha thermal tran r glass transi programmin ent switching developed by d PCL and p olymer netwo g rise to a Tm CLEG materi
ork by heatin
mory (TSM) a nt transition two tempora nd temporary ase-separated nsition.143 Th tion tempera ng at differen g.147
y Lendlein e
poly(cyclohe ork (MACL) m/Tg and Tm/
ial which ha
ng to 40 oC a
allowing for s (Th, Tl), so ary shapes, w y shape throu d domains w he thermal atures in the nt temperatu
t al. using tw exyl
), the second Tm system, s been
21
programmed in two steps. When triggered by heating above T1 (40 oC) it unfolds to recover the 1st temporary shape and upon heating above T2 (60 oC) reforms hanging anchors of the original shape. Similar to dual shape memory development, TSPs have also been made to utilize indirect thermal actuation through nanocomposites containing iron (III) oxide.148 TSM is also present in some LCE systems which also possess shape changing ability through different LC transitions.142
1.6 Conclusions
The previous sections described the current state of SMPs, specifically the extension of triggering to various external stimuli and the development of multifunctional triple shape materials. As discussed, the use of light as a trigger independent of thermal mechanisms is advantageous for biomaterials allowing for remote triggering at ambient temperatures. Although multiple LSMPs have been demonstrated, there are still areas that need to be addressed including uniformity, elasticity, tailoring of properties, cytotoxicity, and extension into multifunctional biomaterials. The following chapters describe in detail efforts to
produce materials with a range of properties which embody the characteristics of degradable, biocompatible, tunable, multifunctional, and responsive systems.
1.7 Dissertation Organization
This dissertation is organized into five parts. Chapter I is a general discussion of shape memory materials and their recent advances in the areas of triggering, biomaterial applications, and triple shape memory. Chapter II discusses the synthesis and
22
23
References
1.) Yoshihito, O.; Matsuda, A. Nature, 1995, 376, 219.
2.) Wache, H.; Hentrich, D.; Wagner, M. J. Mater. Sci. 2003, 14, 109.
3.) Maitland, J.; Metzeger, F.; Schumann, D.; Lee, A.; Wilson, S. Laser Surg. Med. 2002,
30, 1.
4.) Langer, R.; Lendlein, A. World Patent WO 2003088818 A2, 2003.
5.) Gall, K.; Yakacki, C.; Liu, Y.; Shandas, R.; Willett, N.; Anseth, K. J. Biomed. Mater. Res., Part A 2005, 73, 339
6.) Adachi,H.; Yokoi, T.; Hatori, T.; Morishita, K.; Sakashita, K.; Kaiya, H.; Inoue, K.; Ueda, Y.; Nakamura, T.; Yamaguchi, S. Japan Patent 02124438, 1990.
7.) Kobayashi, K.; Hayashi, S. Japan Patent 02183132, 1990. 8.) Kondo, S.; Hayashi, S. Japan Patent 03183920, 1991.
9.) Lan X, Liu, Y.; Lv, H.; Wang, X.; Leng, J.; Du, S. Smart Mater Struct 2009, 18, 240021.
10) Arzberger SC, Munshia NA, Lakea MS, Wintergerst J, Varlese S, Ulmer MP. Optical Materials and Structural Technologies Proceedings of SPIE 2003, 5179, 143.
11.) Benett WJ, Krulevitch PA, Lee AP, Northrup MA, Folta JA.United States Patent 5609, 608, 1997.
12.) Kobayashi K, Hayashi S. United States Patent 5098776; 1992.
13.) Ji F, Zhu Y, Hu J, Liu Y, Yeung L, Ye G. Smart Mater Struct 2006, 15, 1547. 14.) Behl, M.; Zotzmann, J.; Lendlein, A. Adv Polym Sci 2010, 226, 1.
15.) Lendlein, A. Kelch, S. Angew Chem Int 2002, 41, 2034. 16.) Luo X.; Mather P.T. Macromoleucles 2009, 42, 7251. 17.) Chang, L.C.; Read, T.A. J Metals 1951, 191, 47.
18.) Liu, C.; Qin, H.; Mather, P.T. J Mater Chem 2007, 17, 1543.
24
20.) Ota, S. Radiat Phys Chem 1981, 18, 81.
21.) Mondal, S.; Hu, J.L. J Elastom Plast 2007, 39, 81.
22.) Chen, S.; Hu, J.; Liu, Y.; Liem, H.; Zhu, Y.; Liu, Y. J Polym Sci Part B, Polymer Physics 2007, 45, 444.
23.) Cao, Q.; Liu, P. Polym Bull 2006, 57, 889.
24.) Lin, J.R.; Chen, L.W. J Appl Polym Sci 1998, 69, 1563. 25.) Lin, J.R.; Chen, L.W. JAppl Polym Sci 1998, 69, 1575.
26.) Jiangand, H.Y.; Schmidt, A.M. In: Shape-memory polymers and multifunctional composites, CRC Press LLC, 2010, p. 21-63.
27.) Li, F.; Hou, J.; Zhu, W.; Zhang, X.; Xu, M.; Luo, X.; Ma, D.; Kim, B.K. J Appl Polym Sci 1996, 62, 631.
28.) Ikematsu, T.; Kishimoto, Y.; Karaushi, M. Japan Patent 02022355, 1990. 29.) Jeon, H.G.; Mather, P.T.; Haddad, T.S. PolymInt 2009, 49, 453.
30.) Luo, X.; Zhang, X.; Wang, M.; Ma, D.; Xu, M.; Li, F. J Appl Polym Sci 1997, 64, 2433.
31.) Kraft, A.; Rabani, G.; Schuh, C.; Mueller, K.; Lechmann, M.C. Polym Mater Sci Eng
2005, 93, 935.
32.) Hu JL. In: Characterization of shape memory properties in polymers. CRC Press LLC, 2007. p. 197–225.
33.) Yakacki CM, Willis S, Luders C, Gall K. Adv Eng Mater 2008, 10, 112.
34.) Kim BK, Lee SY, Lee JS, Baek SH, Choi YJ, Lee JO, et al. Polymer 1998, 39, 2803. 35.) Leng, J.; Lan, X.; Liu, Y.; Du, S. Prog Mater Sci 2011, 56, 1077
36.) Li, F.; Zhu, W.; Zhang, X.; Zhao C.; Xu, M. J Appl Polym Sci 1999, 71, 1063. 37.) Skakalova, V.; Lukes, V.; Breza, M. Macromol Chem Phys 1997, 198, 3161. 38.) Hu, Z.; Zhang, X.; Li, Y. Science 1995, 269, 525.
25
40.) Zhang, D.; Lan, X. Liu, Y.; Leng, J. In: Behavior and mechanics of multifunctional and composite materials, Proc. SPIE 6526, 2007.
41.) Park, C.; Lee, J.Y.; Chun, B.C.; Chung, Y.C.; Cho, J.W.; Cho, B.G. J Appl Polym Sci
2004, 94, 308.
42.) Hu, J.; Yang, Z.; Yeung, L.; Ji, F.; Liu, Y. Polym Int 2005, 54, 854. 43.) Lee, S.H.; Kim, J.W.; Kim, B.K. Smart Mater Struc 2004, 13, 1345.
44.) Alteheld, A.; Feng, Y.; Kelch, S.; Lendlein, A. Angew Chem Int Ed 2005, 44, 1188. 45.) Yang, Z.; Hu, J.; Liu, Y.; Yeung, L. Mater Chem Phys 2006, 98, 368.
46.) Balazs, A.; Emrick, T.; Russell, T.P. Science 2006, 314, 1107. 47) Manias, E. Nat Mater 2007, 6, 9.
48) Manpreet S. Thermal characterization of nanocomposite shape memory polymer for their mechanical and thermal properties. Thesis of graduate studies, Lamar
University. UMI Number 1429868.
49) Ohki, T.; Ni, Q.Q.; Iwamoto, M. Sci Eng Compos Mater 2004, 11, 137. 50) Zhang, C.S.; Ni, Q.Q. Compos Struct 2007, 78, 153.
51) Liang, C.; Rogers, C.A.; Malafeew, E. J Intelligent Mater Syst Struct 2000, 11, 877. 52) Lan, X.; Huang, W.M.; Leng, J.S.; Liu, Y.J.; Du, S.Y. Adv Mater Res 2008, 47, 714. 53) Wornyo, E.; Gall, K.; Yang, F.; King, W. Polymer 2007, 48, 3213.
54) Sahoo, N.G.; Jung, Y.C.; Yoo, H.J.; Cho, J.W. Compos Sci Technol 2007, 67, 1920. 55) Bhattacharyya, A.R.; Sreekumar, T.V.; Liu, T.; Kumar, S.; Ericson, L.M.; Hauge,
R.H. Polymer 2003, 44, 2373.
56) Assouline, E.; Lustiger, A.; Barber, A.H.; Cooper, C.A.; Klein, E.; Wachtel, E. J Polym Sci Part B 2003, 41, 520.
57) Seoul, C.; Kim, Y.T.; Baek, C.K. J Polym Sci Part B Polym Phys 2003, 41, 1572. 58) Liu, T.; Phang, I.Y.; Shen, L.; Chow, S.Y.; Zhang, W.D. Macromolecules 2004, 37,
26
59) Lou, X.; Detrembleur, C.; Sciannamea, V.; Pagnoulle, C.; Jerome, R. Polymer 2004,
45, 6097.
60) Wong, M.; Paramsothy, M.; Xu, X.J.; Ren, Y.; Li, S.; Liao, K. Polymer 2003, 44, 7757.
61) Gojny, F.H.; Schulte, K. Compos Sci Technol 2004, 64, 2303.
62) Sandler, J.K.W.; Kirk, J.E.; Kinloch, I.A.; Shaffer, M.S.P.; Windle, A.H. Polymer
2003, 44, 5893.
63) Shaffer, M.S.P.; Windle, A.H. Adv Mater 1999,11, 937.
64) Buckley, P.R.; McKinley, G.H.; Wilson, T.S.; Small, W.; Benett, W.J.; Bearinger, J.P. IEEE Trans Biomed Eng 2006, 53, 2075.
65) Razzaq, M.Y.; Anhalt, M.; Frormann, L.; Weidenfeller, B. Mater Sci Eng A – Struct Mater Prop Microstruct Process 2007, 471, 57.
66) Yakacki, C.M.; Satarkar, N.S.; Gall, K.; Likos, R.; Hilt, J.Z. J Appl Polym Sci 2009,
112, 3166.
67) Hazelton, C.S.; Arzberger, S.C.; Lake, M.S.; Munshi, N.A. J Adv Mater 2007, 39, 35. 68) Vaia, R. Nat Mater 2005, 4, 429.
69) Rousseau, I.A.; Mather, P.T. J Am Chem Soc 2003, 125, 15300.
70) Rousseau, I.A.; Qin, H.H.; Mather, P.T. Macromolecules 2005, 38, 4103.
71) Zhang, Q.M.; Li, H.; Poh, M.; Xia, F.; Cheng, Z.Y.; Xu, H.; Huang, C. Nature
2002, 419, 284.
72) Hiraoka, K.; Sagano, W.; Nose, T.; Finkelmann, H. Macromolecules, 2005, 38, 7352. 73) M. Warner and E. M. Terentjev, Liquid Crystalline Elastomers, Oxford University
Press, Oxford, UK, 2003.
74) Lendlein, A.; Schmidt, A.M.; Langer, R. Proc Natl Acad Sci 2001, 98, 842. 75) Brennan, M. Chem Eng News 2001, 79(6), 5.
76) Ashley, S. Sci Am 2001, No. 5.
27
78) Laurencin, C.T.; Sobrasua, I.E.M.; Langer, R.S. in Biomedical Applications of Synthetic Biodegradable Polymers, CRC Press, Boca Raton, 1995.
79) J.D. Bronzino The Biodmedical Engineering Handbook, CRC Press, Boca Raton, 1995.
80) Petrova, P.S.; Manolova, N.; Rashkov, I.; Li, S.; Vert, M. Polym Int 1998, 45, 419. 81) Cohn, D.; Stern, T.; Gonzalez, F.; Epstein, J. J Biomed Mater Res 2002, 59, 273. 82) Liu, L.; Sheardown, H. Biomaterials 2005, 26, 233.
83) Williams, D.F. Sadhana 2003, 28, 563.
84) Wang, W.; Ping, P.; Chen, X.; Jing, X. Eur Polym J 2006, 42, 1240.
85) Mohr, R.; Kratz, K.; Weigel, T.; Lucka-Gabor, M.; Moneke, M.; Lendlein, A. Proc Natl Acad Sci USA 2006, 103, 3540.
86) Weigel, T.; Mohr, R.; Lendlein, A. Smart Mater Struct 2009, 18, 025011.
87) Koerner, H.; Price, G.; Pearce, N.A.; Alexander, M.; Vaia, R.A. Nat Mater 2004, 3, 115.
88) Liu, Y.; Lv, H.; Lan, X.; Leng, J.; Du, S. Compos Sci Technol 2009, 69, 2064. 89) Small, W.; Wilson, T.S.; Benett, W.J.; Loge, J.M.; Maitland, D.J. Opt Express 2005,
13, 8204.
90) Yang, B.; Huang, W.M.; Li, C.; Lee, C.M.; Li, L. Smart Mater Struct 2004, 13, 191. 91) Huang, W.M.; Yang, B.; An, L.; Li, C.; Chan, Y.S. Appl Phys Lett 2005, 86, 114105. 92) Yang, B.; Huang, W.M.; Li, C.; Li, L. Polymer 2006, 47, 1348.
93) Yang, B.; Huang, W.M.; Li, C.; Li, L.; Chor, J.H. Scr Mater 2005, 53, 105
94) Biercuk, M.J.; Llaguno, M.C.; Radosavljevic, M.; Hyun, J.K.; Johnson, A.T.; Fischer, J.E. Appl. Phys. Lett. 2002, 80, 2767.
95) Chang, T.E.; Jensen, L.R.; Kisliuk, A.; Pipes, R.B.; Pyrz, R.; Sokolov, A.P. Polymer
2005, 46, 439.
96) Gall, K.; Dunn, M.L.; Liu, Y; Finch, D.; Lake, M.; Munshi, N.A. Acta Mater 2002,
28
97) Leng, J.S.; Lan, X.; Liu, Y.J.; Du, S.Y. Smart Mater Struct2009, 18, 074003. 98) Leng, J.S.; Lan, X.; Liu, Y.J.; Du, S.Y.; Huang, W.M.; Liu, N. Appl Phys Lett
2008, 92, 014104.
99) Leng, J.S.; Lv, H.B.; Liu, Y.J.; Du, S.Y. Appl Phys Lett 2007, 91, 144105. 100) Leng, J.S.; Lv, H.B.; Liu, Y.J.; Du, S.Y. J Appl Phys 2008, 104, 104917.
101) Cho, J.W.; Kim, J.W.; Jung, Y.C.; Goo, N.S. Macromol Rapid Commun 2005, 26, 412.
102) Razzaq, M.Y.; Anhalt, M.; Frormann, L.; Weidenfeller, B. Mater Sci Eng A – Struct Mater Prop Microstruct Process 2007, 444, 227.
103) Schmidt, A.M. Macromol Rapid Commun 2006, 27, 1168. 104) Zhang, H.; Xia, H.; Zhao, Y. J Mater Chem 2012, 22, 845.
105) Leng, J.; Zhang, D.; Liu, Y.; Yu, K.; Lan, X. Appl Phys Lett 2010, 96, 111905. 106) Leng JS, Wu XL, Liu YJ. Appl Polym Sci 2009, 114, 2455.
107) Behl, M.; Lendlein, A. Mater Today 2007, 10, 20.
108) Leng, J.S.; Lv, H.B.; Liu, Y.J.; Du, S.Y. Appl Phys Lett 2008, 92, 206105. 109) Lv, H.B.; Leng, J.S.; Liu, Y.J.; Du, S.Y. Adv Eng Mater 2008, 10, 592. 110) Kumpfer, J.R.; Rowan, S.J. J Am Chem Soc 2011, 133, 12866.
111) Jiang, H.Y.; Kelch, S.; Lendlein, A. Adv Mater 2006, 18(11), 1471. 112) Irie, M. Adv Polym Sci 1990, 94, 27.
113) Brown, G.H. Photochromism 1971, Wiley-Interscience. 114) Lovrien, R. PNAS 1967, 57, 236.
115) Irie, M.; Suzuki, T. Macromol Chem Rapid Commun 1981, 8, 607. 116) Irie, M.; Hosoda, M. Macromol Chem Rapid Commun 1985, 6, 533. 117) Irie, M.; Iga, R. Macromol Chem Rapid Commun 1987, 8, 569.
29
119) Kumar, G.S.; DePra, P.; Zhang, K.; Neckers, D.C. Macromolecules 1984, 17, 2463. 120) Kumar, G.S.; Savariar, C.; Sattran, M.; Neckers, D.C. Macromolecules 1985, 18,
1525.
121) Zimmerman, E.K.; Stille, J.K. Macromolecules, 1985, 14, 1246. 122) Irie, M.; Menju, A.; Hayashi, K. Macromolecules, 1981, 12, 1176. 123) Yu, Y.L.; Nakano, M.; Ikeda, T. Nature 2003, 425, 145.
124) Camacho-Lopez, M.; Finkelmann, H.; Palffy-Muhoray, P.; Shelley, M. Nat Mater
2004, 3, 307.
125) Ikeda, T.; Nakano, M.; Lei, Y.Y.; Tsutsumi, O.; Kanazawa, A. Adv Mater 2003, 15, 307.
126) Finkelmann, H.; Nishikawa, E.; Pereira, G.G.; Warner, M. Phys Rev Lett 2001, 87, 015501.
127) Li, M.H.; Keller, P.; Li, B.; Wang, X.G.; Bruner, M. Adv Mater 2003, 15, 569. 128) Irie, M.; Kunwatchakun, D. Macromolecules 1986, 19, 2476.
129) Mamada, A.; Tanaka, T.; Kunwatchakun, D.; Irie, M. Macromolecules 1990, 23, 1517.
130) Suzuki, A.; Tanaka, T. Nature 1990, 346, 345.
131) Juodkazis, S.; Mukai, N.; Wakaki, R.; Yamaguchi, A.; Matsuo, S.; Misawa, H.
Nature 2000, 408, 178.
132) Hogan, P.M.; Tajbakhsh, A.R.; Terentjev, E.M. Phys Rev E: Stat Nonlinear Soft Matter Phys 2002, 65, 041720.
133) Yu, Y.L.; Nakano, M.; Shishido, A.; Shiono, T.; Ikeda, T. Chem Mater 2004, 16, 1637.
134) Yu, Y.L.; Ikeda, T. Macromol Chem Phys Suppl 2005, 206, 1705. 135) Kumar, G.S.; Neckers, D.C. Chem Rev 1989, 89, 1915.
30
137) Nakano, M.; Yu, Y.; Shishido, A.; Tsutsumi, O.; Kanazawa, A.; Shiono, T.; Ikeda, T.
Mol Cryst Liq Cryst 2003, 398, 1.
138) Yu, Y.; Nakano, M.; Ikeda, T. Pure Appl Chem 2004, 76, 1435.
139) Lee, K.M.; Koerner, H.; Vaia, R.A.; Bunning, T.J.; White, T.J. Soft Matter 2011, 7, 4318.
140) Lendlein, A.; Jiang, H.Y.; Junger, O.; Langer, R. Nature 2005, 434, 879. 141) Wu, L.; Jin, C.; Sun, X. Biomacromolecules 2011, 12, 235.
142) Behl, M.; Lendlein, A. J. Mater. Chem. 2010, 20, 3335.
143) Bellin, I.; Kelch, S.; Langer, R.; Lendlein, A. Proc Natl Acad Sci 2006, 103(48), 18043.
144) Pretsch, T. Smart Mater. Struct. 2010, 19, 1.
145) Chen, S.; Hu, J.; Yuen, C.H.; Chan, L.; Zhuo, H. Polym Adv Technol 2010, 21, 377.
146) Behl, M.; Bellin, I.; Kelch, S.; Wagermaier, W.; Lendlein, A. Adv. Funct. Mater.
2009,19,102.
147) Xie, T. Nature Letters 2010, 464, 267.
Chapter II
PHOTO-RESPONSIVE POLY(ESTER URETHANE)S FOR DUAL SHAPE MEMORY BIOMATERIALS
2.1 Introduction
Shape memory polymers (SMPs) are proving to be promising enabling materials in the field of minimally invasive implants and smart biomedical devices.1,2 SMPs are
32
Scheme 2.1 Photo-induced reversible [2+2] cycloaddition of two cinnamate functional groups.
Several researchers have described shape changes induced by light that are
independent of heat,10,11,12 however, the most closely related to the present work is based on moieties which undergo a reversible UV induced [2+2] cycloaddition (Scheme 2.1).
Lendlein et al. programmed an acrylate-based hydrogel containing cinnamate groups into fixed shapes by ultraviolet light illumination (λ > 260 nm). When exposed a second time to a higher energy ultraviolet light (λ < 260 nm), newly formed temporary crosslinks were cleaved and the polymer network recovered its original shape, demonstrating that remote light activation at ambient temperatures was possible.13 Figure 2.1 shows an illustration of this programming and recovery process. More recently, Wu et al. utilized polyester
segments functionalized with similar photo-responsive groups randomly distributed them in a thermoplastic polyurethane system to create materials that were hydrolytically degradable.14 For such materials the strain fixity is much lower when compared with thermal shape
33
SME has also been shown in systems based on the photochromic reaction of azobenzene15, however, these materials lack a degradable backbone and other properties advantageous for biomaterials.
For shape memory materials in biomedical applications, targeting thermal transitions can be challenging, and little flexibility for tailoring other properties remains once Ttrans has been set. The use of a light-induced mechanism removes dependence on a thermal transition and allows switching at ambient temperatures. Herein, we present the synthesis and
characterization of a library of biodegradable amorphous poly(ester urethane) prepolymers which are functionalized with a photo-responsive group in each repeat unit to give a uniform
34
distribution along the polymer backbone. The thermoset networks that result from thermal curing possess a range of thermal and mechanical properties as well as the ability to undergo light induced shape memory.
2.2 Experimental Section
2.2.1 Materials
All reagents were purchased from Sigma-Aldrich and used without further
purification unless otherwise noted. Dichloromethane was dried by distilling from CaSO4. Triethylamine and pyridine were dried by distilling from CaH2. 1,8-octanediol was
recrystallized from THF. 2.2.2 Instrumentation.
Gel permeation chromatography was used to determine molecular weights and molecular weight distributions, Mw/Mn, of polymer samples using a Waters Alliance 2695 and Waters 2414 Refractive Index detector. Molecular weights were calculated using a calibration plot constructed from polystyrene standards (Polyscience Corp.). The
35 2.2.3 Monomer Synthesis
Synthesis of Diethyl 2,2'-(Cinnamoylazanediyl)diacetate (DCA) (1). A solution of cinnamoyl chloride (4.165 g, 25.0 mmol) in dry CH2Cl2 was added dropwise into a stirring solution of diethyl iminodiacetate (4.57 mL, 25.5 mmol), triethylamine (7.02 mL, 50.5 mmol) and dry CH2Cl2 (115 mL) at 0 oC. The reaction solution was allowed to stir for 30 minutes at 0 oC and then warmed to room temperature and left stirring for 12 h. The
precipitant was removed by gravity filtration and the solution was washed sequentially with 1.0 M HCl, saturated NaHCO3, and DI H2O. The organic layer was dried over MgSO4, filtered, and excess solvent was removed under reduced pressure. The crude product was recrystallized from hot CH2Cl2/hexanes and dried under vacuum to afford a white solid in 93% yield. 1H-NMR: δ (ppm) = 7.67 (m, 2H), 7.64 (d, 1H, J = 15.3 Hz), 7.41 (m, 3H), 7.14 (d, 1H, J = 15.6 Hz), 4.55 (s, 2H), 4.28 (s, 2H), 4.20 (q, 2H, J = 7.1 Hz), 4.16 (q, 2H, J = 7.1 Hz), 1.25 (t, 6H, J = 7.2 Hz).
Synthesis of Diethyl 3-(Cinnamoyloxy)pentanedioate (DCE) (2). A solution of cinnamoyl chloride (10 g, 60 mmol) in dry CH2Cl2 was added dropwise into a stirring
solution of diethyl 3-hydroxygluturate (10.21 g, 50 mmol) and dry CH2Cl2 (100 mL) at 0 oC. After the addition was complete, pyridine (4.83 mL, 60 mmol) was added dropwise. The reaction solution was allowed to stir for 30 minutes at 0 oC and then warmed to room
36
16 Hz), 5.62 (qn, 1H, J = 6.0 Hz) 4.11 (q, 4H. J = 7.3 Hz), 2.80 (dd, 4H, J1 = 3.8 Hz, J2 = 4.4 Hz) 1.21 (t, 6H, J = 6.0 Hz).
Synthesis of N,N-bis(2-hydroxyethyl)cinnamamide (CAD) (3). A solution of diethanolamine (2.00 g, 19 mmol) and Et3N (9.31 mL, 67 mmol) in dry CH2Cl2 under N2 atmosphere was cooled to 0 oC in an ice bath. To this mixture a solution of cinnamoyl chloride (9.51 g, 57 mmol) in dry CH2Cl2 was added dropwise over a period of 20 min. After complete addition the reaction was allowed to stir for 1 h before removal of the ice bath and returning the reaction to room temperature to stir overnight. The solvent was then
removed by rotary evaporation and methanol was added to dilute the residue. Saturated Na2CO3 was added and the solution was stirred for 3 h before being diluted with DI water. After an extraction with CH2Cl2, the organic layer was dried over MgSO4 and the product was concentrated by vacuum evaporation. Flash chromatography (10:1 CHCl3/MeOH) followed by vacuum drying resulted in a white powder in 68% yield. 1H-NMR: δ (ppm) = 7.66 (d, 1H, J = 16 Hz), 7.50 (m, 2H), 7.35 (m, 3H), 6.94 (d, 1H, J = 16 Hz), 3.88 (dt, 4H, J1 = 6 Hz, J2 = 16 Hz) 3.70 (bs, OH), 3.66 (q, 4H, J = 5.3 Hz).
2.2.4 Polyester Prepolymer Synthesis
Polymerization with DCA Monomer (4, 5). Prepolymers were synthesized with either
37
reduced to 40 torr, after another 5 hours the pressure reduced to 20 torr, and after 18 hours the pressure was reduced to 0.1 torr. The mixture continued to stir under reduced pressure for another 24 hours, to give a total of 48 hours reaction time. The reaction was removed from heat and atmospheric pressure was re-established. The polymer was dissolved in 5 mL of chloroform and precipitated into cold stirring methanol (-78 oC). The polymer was then dried under vacuum for 24 hours. 1H-NMR: δ (ppm) = 7.66 (m, 2H), 7.65 (d, 1H, J = 15.2 Hz), 7.40 (m, 3H), 7.14 (d, 1H, J = 15.2 Hz), 4.56 (s, 2H) 4.29 (s, 2H) 4.13 (t, 2H, J = 5.4 Hz), 4.10 (t, 2H J = 5.4 Hz), 3.52 (t, 0.08H, J = 5.8 Hz) 1.61 (m, 4H) 1.29 (m, 8H).
Polymerization with DCE Monomer (6, 7). An example of a typical polymerization with the DCE monomer is given. A three-necked round bottom flask was charged with monomer 2 (1.663 g, 4.97 mmol), 1,8-octanediol (0.733 g, 5.01 mmol), and Lipase acrylic resin catalyst (240 mg, 10 wt% monomers), evacuated and filled with N2. The mixture of monomers and catalyst is heated to 80 oC to form a melt and stirring is begun. After 2 hours, pressure is slowly reduced to 40 torr for 12 hours, then reduced to 20 torr for 24 hours, and finally reduced to 0.1 torr for a final 12 hours. Heat is removed and atmospheric pressure restored, followed by the polymer being dissolved in 5 mL of chloroform and precipitated into cold stirring methanol (-78 oC). The polymer was collected and dried under vacuum for 24 hours. 1H-NMR: δ (ppm) = 7.66 (d, 1H, J = 16.0 Hz), 7.50 (m, 2H), 7.38 (m, 3H), 6.38 (d, 1H, J = 16.0 Hz), 5.64 (qn, 1H, J = 6.0 Hz), 4.05 (m, 4H), 3.74 (m, endgroup), 2.79 (d, 4H, J = 4.0 Hz), 1.85 (m, endgroup), 1.57 (m, 4H), 1.23 (m, 8H).
Polymerization with CAD Monomer (8). Polymerization with diol monomer 3
required the use of either a complementary diacid or diester, therefore,
38
Figure 2.2 Bifunctional monomers DCA, DCE, CAD and resulting prepolymers from polycondensation with complementary monomers.
dried round bottom flask was charged with monomer 3 (3.13 g, 13.3 mmol) and
1,4-cyclohexanedicarboxylic acid (2.22 g, 12.9 mmol). After purging with nitrogen for several minutes, the vessel was heated to 125 oC to form a melt solution, afterwhich tin (II)
39
Hz), 4.28 (m, 4H), 3.76 (m, 4H), 2.48 (s, 2H), 2.25 (s, 2H), 2.04 (m, 4H), 1.86 (m, 4H), 1.61 (m, 4H), 1.44 (m, 4H).
2.2.5 Thermoset Network Formation
Prepolymer end-functionalization. Hydroxy-terminated polymer of known
M
n(43 mg, 6.78 x 10-3 mmol) was dissolved in CH2Cl2 (2.0 mL). The solution was heated to reflux at 60 oC, then 2-isocyanatoethyl methacrylate (5 µL, 0.035 mmol) was added by syringe. One drop of stannous octoate was also added to help catalyze the reaction. The solution was left under reflux for 2 h, excess solvent was removed under reduced pressure, and the concentrated solution was precipitated in cold stirring methanol (-78 oC). The resulting polymer was transferred and dried under vacuum for 1 day. Example for DCA-ODprepolymer, 1H-NMR: δ (ppm) = 7.65 (m, 2H), 7.64 (d, 1H, J = 15.2 Hz), 7.39 (m, 3H), 7.13 (d, 1H, J = 15.6 Hz), 6.09 (s), 4.55 (s, 2H), 4.28 (s, 2H), 4.11 (m, 4H), 3.97 (q), 3.43 (q), 1.90 (s), 1.60 (m, 4H), 1.28 (m, 8H).
Crosslinking. Prepolymers with crosslinkable endgroups were thermally cured by
40
stirring for 5 minutes, the solution was cast in the mold or on the glass slide, solvent evaporated, and placed in the oven at 80 oC overnight.
2.2.6 Characterization
2.2.6.1 Light-induced Shape Memory (LSM)
Films of 0.5-2.0 mm thickness were placed in a specially designed vise apparatus to stretch and hold the sample in a stressed state while irradiating with UV light. Film samples were elongated to a specified length and the entire apparatus placed in a UVP CL 1000 Crosslinker with five 8 watt mercury bulbs (302 nm) at a distance of ~3 cm for a prescribed time. After UV fixing the film sample was removed from the vise to give the temporary shape. The film sample was then irradiated for a prescribed time in the UV chamber with 254 nm bulbs for a prescribed time under no external force. Dimension measurements before stress was applied, in the stressed state, after fixing, and after recovery were taken with a Mitutoyo ABSOLUTE Digimatic 500 Series caliper in triplicate. Strain fixity (Rf) and strain recovery (Rr) were calculated using the following equations,
100 ) ( × − − = = i p i f m u f l l l l N R ε ε 100 ) 1 ( ) ( × − − = − − − = i p r p p m p m r l l l l N N R ε ε ε ε
2.2.6.2 Physiological Degradation and Water Uptake
Degradation studies were performed for the DCA-OD and DCE-CHDM series. Elastomer films of known weight (30-50 mg) were placed in 1 mL of 0.01 M pH 7.4
41
measured. Each measurement was performed on three separate samples. Mass loss (ML) was calculated according to the following equation,
100 × − = i f i m m m ML
Water uptake (WU) was measured for elastomer films placed in 0.01 M pH 7.4 PBS solution at 37 oC for prescribed intervals using the equations,
100 × − = d d sw m m m
WU ( )= − ×100
i i sw m m m Abs WU
where msw, md, and mi represent the swollen mass, the dry mass, and the initial mass,
respectively. Film samples of known weight were removed from PBS and blotted dry before weighing swollen mass, followed by drying under vacuum for 1 day to obtain the dry mass. 2.2.6.3 Cytotoxicity Studies
Initial cytotoxicity studies were performed by ATP assay using the HeLa cell line. Polymer material was placed in well plates containing cells and cell medium. Plates were incubated at 37 oC in the dark for 3 days, after which the percent viability was found using a CellTiter-Glo® luminescent cell viability kit which determined the amount of
bioluminescent ATP present in cells. Each measurement was done in triplicate for crosslinked DCA-OD materials.
42 2.3.1 Monomer Synthesis
The successful synthesis of both diester monomers (DCA, DCE) and diol monomer (CAD) was achieved by reacting the acyl chloride derivative of cinnamic acid, with diethyl iminodiacetate, 3-hydroxygluturate, or diethanolamine at low temperatures (0 oC) (Scheme 2.2). Due to the high reactivity of acid chlorides, low temperatures were used in order to decrease reactivity and to control the heat generated. Triethylamine (Et3N) was employed for the synthesis of DCA (1) and CAD (3) but could not be used for the DCE (2) synthesis due to the difference in nucleophilicity of the 2o amine and the 1o alcohol. Due to the nature of the less nucleophilic alcohol, Et3N inhibits the reaction with cinnamoyl chloride because in the reaction mixture it is the most nucleophilic species. Pyridine, whose stabilized ring structure decreases its nucleophilicity, was employed instead.
43 Scheme 2.2 Bifunctional photo-monomer synthesis.
corresponding to the cis-methylene and one to the trans-methylene with respect to the carbonyl. The restricted rotation is also evident in the overlapping of the other two methylene signals. Although these protons are not adjacent to the amide bond, they still encounter different magnetic environments. The same inequivalence can be seen in the 1 H-NMR of CAD (3), however, it is not as substantial due to the lower molecular weight of the substituents on the amine of the amide bond.
In DCE (2) the rotational barrier around the ester linkage is much smaller in
44
45
will help increase the probability of cinnamate groups interacting in the solid state network in order to undergo the photo induced [2+2] cycloaddition.
2.3.2 Polymer Synthesis
The successful polycondensation of DCA with various diols was achieved by bulk melt polymerization with tin octoate catalyst, high temperatures (130 oC), and reduced pressures (0.1 atm). Scheme 2.3 (top) shows the reaction of DCA with 1,8-octanediol. The high reaction temperatures and reduced pressure was utilized to remove the ethanol
condensate which is necessary to push the reaction to high conversion. The high temperature was also necessary to activate the catalyst. Tin octanoate (SnOct) in turn activates the ester group of the monomer for transesterification through coordination. Tin catalyzed
polycondensation of DCE was not successful due to the transesterification of the cinnamate group during polymerization, which led to branching and loss of photo-groups. An
enzymatic catalyst (Novozym 435-Lipase) was used instead for all polymerizations involving DCE. Novozym 435-Lipase catalyst is known to be directed towards primary alcohols or esters, thereby not affecting the photo-groups, and leading to linear polymer with no cleavage of photo groups. Scheme 2.3 (middle) shows the reaction of DCE with 1,4-cyclohexanedimethanol.
46 Scheme 2.3 Polymerization of polyester prepolymers.
used to target molecular weights based on a calculated imbalance. This imbalance uses the diol in excess, thereby controlling the chain ends and giving hydroxyl-terminated polymers.
Unsaturated endgroups were successfully added to the polymer chain ends via the reaction of hydroxyl-terminated polyesters with 2-isocyanatoethyl methacrylate in CH2Cl2 at reflux for 2 h. These cross-linkable unsaturated ends can be thermally initiated by a free radical initiator AIBN, reacted with a thiol crosslinker through a thiol-ene method, or photo-initiated, however, there is the likelihood of the latter affecting the photo-responsive moieties in an undesired manner. Therefore only the two thermal methods were utilized to form thermoset networks.
2.3.3 Thermal and Mechanical Analysis
47
permeation chromatography show good correlation with stoichiometric imbalance to target polymer chain length and endgroups. A wide range of glass transition temperatures (Tg) for the completely amorphous prepolymers were seen, with the cyclic diol monomer restricting rotation and giving rise to higher glass transitions (~30 oC higher Tg than DCA-OD). The linkage chemistry between the polymer backbone and the pendant photo-group also affected the thermal properties. Comparing prepolymers containing the same diol monomer (linear or cyclic), those possessing cinnamamide linkages generally saw an increase of Tg by ~40 oC over those with cinnamate linkages. This is again associated with the rotational barriers around the different linkage bonds. Higher thermal decomposition temperatures were also
Table 2.1 Molecular weight and thermal characterization of photo-responsive prepolymers.
Decomposition Temp.b
Polymer Sample n
M a (g/mol x 10
-w
M /Mn
a 5% (oC) 10% (oC) Tg (oC)c
DCA-OD-5 5.4 1.6 301 319 4
DCA-OD-8 7.9 1.8 371 395 6
DCA-CHDM-5 4.9 1.9 365 382 35
DCA-CHDM-8 7.2 1.4 343 414 43
DCE-OD 1.9 2.1 248 268 -34
DCE-CHDM 2.7 1.6 293 306 -7
48
Table 2.2 Thermal and mechanical properties of photo-responsive thermoset networks.
Decomposition
Series Prepoly MW (g/mol)a 5% (oC) 10% (oC) Tg (oC)c G (MPa)d Breakd
DCA-CHDM 7000 322 355 52 61.0 5 %
DCA-OD 8000 366 392 20 28.0 32 %
DCE-CHDM 3000 300 319 7 15.7 90 %
DCE-OD 2000 267 340 -13 0.4 330 %
aGPC- PS Stds, bTGA, cDSC, dInstron (23 oC)
seen for amide containing prepolymers synthesized with either DCA or CAD monomers. Having these two handles to alter thermal properties, as well as the impact of varying molecular weights, allows for the creation of a range of material properties.
The amorphous nature of the prepolymers made for easier processing to form permanent networks of desired size and shape by molding or solvent casting. Table 2.2 shows the thermal and mechanical properties of the resulting thermoset networks. As was expected the Tg increased from prepolymer to thermoset network by about 15 oC and again, a large range of glass transition temperatures is seen which in turn produces a range of
mechanical properties as characterized by Instron at room temperature (23 oC). DCA-CHDM
exhibited the highest Young’s modulus (61 MPa) with very small elongation (5%)