Genome-wide association study in Guillain-Barré syndrome
Stefan Blum, Ying Ji, David Pennisi, Zhixiu Li, Paul Leo, Pamela McCombe, Matthew A. Brown
PII: S0165-5728(18)30033-X
DOI: doi:10.1016/j.jneuroim.2018.07.016
Reference: JNI 476822
To appear in: Journal of Neuroimmunology
Received date: 23 January 2018 Revised date: 27 July 2018 Accepted date: 27 July 2018
Please cite this article as: Stefan Blum, Ying Ji, David Pennisi, Zhixiu Li, Paul Leo, Pamela McCombe, Matthew A. Brown , Genome-wide association study in Guillain-Barré syndrome. Jni (2018), doi:10.1016/j.jneuroim.2018.07.016
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCEPTED MANUSCRIPT
Genome-wide Association Study in Guillain-Barré Syndrome
Stefan Bluma,1, Ying Jib, c,1, David Pennisib, Zhixiu Lib, Paul Leob, Pamela, McCombea,2 and Matthew A. Brownb,2
1 These authors contributed equally. 2 These authors contributed equally.
aThe University of Queensland, UQ Centre for Clinical Research, Royal Brisbane & Women's Hospital, Brisbane, Australia.
bTranslational Genomics Group, Institute of Health and Biomedical Innovation, School of Biomedical Sciences, Queensland University of Technology (QUT) at Translational Research Institute, Brisbane, Australia.
cDepartment of Neurology, Peking University Third Hospital, Beijing, P. R. China.
*Correspondence to:
Professor Matthew A. Brown, Translational Genomics Group,
Institute of Health and Biomedical Innovation, School of Biomedical Sciences,
Queensland University of Technology (QUT) at Translational Research Institute, Brisbane, Australia
Telephone: +61 7 34437017 Fax: +61 7 34437099 Matt.brown@qut.edu.au
ACCEPTED MANUSCRIPT
ABSTRACT
Guillain-Barré syndrome (GBS) is considered to have an immune-mediated basis, but the genetic contribution to GBS is unclear. We conducted a GWAS involving 215 GBS patients and 1,105 healthy controls. No significant associations of individual SNPs or imputed HLA types were observed. We performed a genome-wide complex trait analysis for evaluation of the heritability of GBS, and found that common SNPs contribute up to 25% of susceptibility to the disease. Genetic risk score analysis showed no evidence of overlap in genetic susceptibility factors of GBS and multiple sclerosis. Given the unexplained heritability of the trait further larger GWAS are indicated.
KEYWORDS: Guillain-Barré syndrome; case-control study; genome-wide association study; heritability; genetic risk score; human leukocyte antigen (HLA) genes.
ACCEPTED MANUSCRIPT
1. INTRODUCTION
Guillain-Barré syndrome (GBS) is a severe acute ascending peripheral neuropathy causing weakness of the limbs. GBS with weakness can be subdivided into acute inflammatory demyelinating polyradiculoneuropathy (AIDP), acute motor axonal neuropathy (AMAN), and acute motor and sensory axonal neuropathy (AMSAN). There are also disorders that are regarded as GBS variants, such as Miller Fisher syndrome (MFS) or focal variants of GBS; [1]. The pathogenesis of GBS is incompletely understood. It is considered to be mediated by immune-mediated
responses, triggered by infection, with two-thirds of patients experiencing a preceding bacterial or viral infection [2, 3]. However, unlike classical autoimmune diseases, GBS is typically monophasic, and is more common in males, and does not have a clear HLA association, as required by the Rose and Bona criteria for autoimmune diseases [4].
GBS has pathological similarities to multiple sclerosis (MS), another inflammatory demyelinating neurological disease. However, in MS the central rather than peripheral nervous system is targeted [5]. MS is a highly heritable disease, found to be
associated with more than 140 genetic variants [6]. A potential genetic basis for GBS, however, is less clear. No systematic twin study has been performed. Increased familiality of the disease has been reported, as well as evidence of anticipation with younger generations developing disease at an earlier age, suggesting a genetic mechanism [7-10].
Many candidate gene studies have been performed in GBS, most focusing on human leukocyte antigen (HLA) genes, the most important genetic regulators of the immune system, as well as other genes [11]. HLA genes are encoded within the Major
ACCEPTED MANUSCRIPT
the regulation of immunity and infection, and is closely linked with most autoimmune diseases [12]. However, to date, most studies of HLA associations with GBS have been negative [13-15], with no positive findings being consistently replicated between studies. A recent meta-analysis has suggested association of TNF genetic variants with disease, but this locus is known to be markedly affected by population stratification, which was not controlled for in the meta-analysis [16]. Similarly, multiple association studies of non-MHC candidate genes have been reported but none has achieved definitive levels of association, nor been consistently replicated. None of these studies has controlled for population stratification, and all have been modest in size. Therefore, we conducted the present genome-wide association study (GWAS) to explore the possible genetic basis of GBS more extensively.
ACCEPTED MANUSCRIPT
2. METHODS
2.1 Demographics
A total of 215 patients with confirmed diagnosis of GBS were included in our study (Table 1). GBS was defined according to the National Institute of Neurological Disorders and Stroke criteria [17], and the majority of patients had level 1 or level 2 certainty of diagnosis according to the Brighton criteria [18](Table 1). 1,105 samples were collected as control group. All samples came from centres in Brisbane,
Townsville, Sunshine Coast and Sydney, Australia. Both case and control subjects were of Caucasian ethnicity. Informed written consent was obtained from all participants. This study was approved by the Human Research Ethics Committee of the Royal Brisbane and Women's Hospital (approval number HREC/QRBW/31).
2.2 Quality control and association analysis
Genotyping was performed in an ISO15189-accredited clinical genomics facility, Australian Translational Genomics Centre (ATGC), Queensland University of Technology. All samples were genotyped by Illumina HumanOmniExpress
(OmniExpress) BeadChip. Quality control (QC) was performed using the PLINK 1.9 package (https://www.cog-genomics.org/plink2)[19, 20] and Shellfish
(http://www.stats.ox.ac.uk/~davison/software/shellfish). Detailed information regarding QC filtering is described below (Table 2). We included all 22 autosomal chromosomes in our study. For individuals, samples with a missing genotype rate higher than 10% or extreme heterozygosity (±3SD from the mean) were excluded. Pairwise identity by descent (IBD) was used to detect cryptic relatedness, with
samples with an IBD > 0.185 excluded from further analysis. For markers, SNPs with a call rate less than 95% or with significant differences (P < 10−5) in missing genotype rate between cases and controls were removed. SNPs identified with extensive
ACCEPTED MANUSCRIPT
frequency (MAF) of <5% were excluded. Differential ethnicity was detected by principal component analysis (PCA) using Shellfish (Supplementary Figure S1). After merging with HapMap genotypes, samples not clearly consistent with a Caucasian ethnicity were excluded. Then a second PCA was conducted to identify outliers, defined as more than ±6SD from the mean for the first principal component, to correct for population stratification. After QC steps were performed, the genotype-phenotype association was tested using logistic regression analysis with the first principal
component as the covariant using PLINK. Genomic inflation was assessed by quantile-quantile (Q-Q) plot and the genomic inflation factor, λ1000.
2.3 Imputation of SNPs in the human leukocyte antigen region
SNPs in the HLA region were specifically imputed by the SNP2HLA software [21], with The National Institute of Diabetes and Digestive and Kidney Diseases Type 1 Diabetes Genetics consortium dataset
(https://www.niddkrepository.org/studies/t1dgc/) used as the reference panel. The logistic regression analysis was performed with dosage data after removing poorly imputed markers and sample outliers. All analyses were performed following the standard procedures according to the SNP2HLA instructions
(http://broadinstitute.rog/mpg/snp2hla/).
2.4 Estimation of the narrow-sense heritability (h2)
Genome-wide Complex Trait Analysis (GCTA) was applied to estimate the variance explained by all the SNPs [22]. Since we were conducting a case-control study, we performed a further, more stringent, QC for this analysis. Markers with a call rate < 95%, or individuals with a genotype missing rate > 1%, were excluded, and 0.05 was chosen as the cutoff for IBD and the P-value for HWE [23]. Genetic relationship matrix (GRM) from all autosomal SNPs was assessed to remove samples with cryptic relatedness (0.025 as the cutoff value), followed by h2 estimation.
ACCEPTED MANUSCRIPT
2.5 Genetic risk score test
A genetic risk score (GRS) test was performed with a set of genome-wide significant multiple sclerosis (MS)-associated SNPs. From published MS studies in the
NHGRI-EBI GWAS catalog [24], we selected a set of SNPs that reached
genome-wide significance for association with MS (P < 5 × 10−8; 49 SNPs at the time of analysis), and compared the allele frequency of these SNPs between cases and controls in our GBS study using both unweighted and weighted genetic risk scores.
ACCEPTED MANUSCRIPT
3. RESULTS
We started with genotype data of 1,320 individuals (215 cases and 1,105 controls) and 699,609 SNPs. After QC, 830 individuals (191 cases and 639 controls) and 579,499 SNPs remained. As shown in the quantile-quantile plot (Supplementary Figure S2), the inflation factor was in the acceptable range, with λ1000 = 1.056 when correcting for one principal component. No SNPs reached the suggestive association threshold (P < 10−5). The three most associated SNPs were rs9371600 (chromosome 6q25, P = 1.56 × 10−5), rs16904331 (chromosome 8q24; P = 1.65 × 10−5) and rs2243523
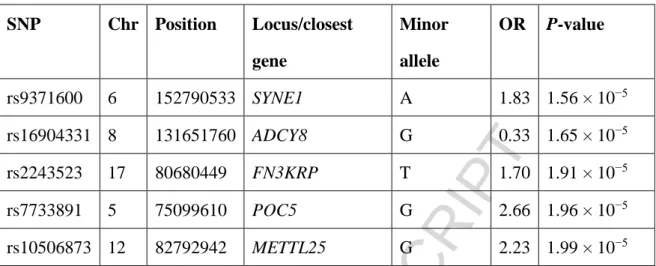
(chromosome 17q25; P = 1.906 × 10−5; Figure 1). Detailed information for the top five associated SNPs is shown in Table 3. Considering directly genotyped SNPs, the strongest MHC association was with SNP rs1264622 (P = 4.37 × 10−3).
3.1 Imputation of HLA genes
Given the previous reported associations of GBS with HLA alleles, we investigated these and the MHC region more closely. A total of 8,961 SNPs within the MHC region, HLA alleles and their composite amino acids were imputed with SNP2HLA. We excluded markers with r2 < 0.5 and samples with unreliable imputed data, leaving 8,597 SNPs and 821 individuals for further logistic regression analysis. No MHC SNPs or imputed HLA alleles reached suggestive significance levels for association with GBS. The strongest SNP association with rs6928738 (P = 2.19 × 10−4; within the MHC Class I region, nearest gene being the hypothetical gene, LOC646570). The strongest HLA association was with HLA-A*3001 (P = 1.29 × 10−2). A recent
meta-analysis suggested that a polymorphism in the TNFA promoter (TNF-α 308G/A, rs1800629, risk allele=A) was associated with GBS . This association was restricted to Asian studies with no association observed in Caucasians, and was only seen with comparison of AA vs GG genotypes or a dominant model (AA vs AG+GG) [16]. No association was observed with allelic analysis (A vs G), or the typical comparison of genotype counts (AA/AG/GG in cases vs controls). In the current study, nominal
ACCEPTED MANUSCRIPT
association was seen with a recessive model (P=0.031) and comparison of AA vs GG genotypes (P=0.027) but no association was seen with either dominant (P=0.23), genotypic (P=0.076) or allelic models (OR= 1.3, risk allele=A, P = 0.076).
3.2 Estimation of the heritability of Guillain-Barré syndrome
As we were unable to identify any variants that were associated with GBS at a genome-wide level of significance, we employed GCTA to estimate the genetic influence on risk of the disease. After filtering with the more stringent QC steps required for case-control studies, 756 individuals and 544,077 SNPs were available for subsequent analysis. h2 estimates were calculated for a range of GBS prevalences (Figure 2). This showed that h2 captured by these SNPs was ≥0.15 for prevalences >1/10,000.
3.3 There is no evidence of a shared genetic basis between Guillain-Barré syndrome and multiple sclerosis
Since GBS and MS have pathological similarities, we sought to determine whether there was a potential shared genetic basis between the two diseases. Therefore, we conducted a GRS test for GBS with a set of MS-associated SNPs with genome-wide significance. A 49-locus genetic risk score derived from MS studies (Supplementary Table S1) was applied in our dataset for allele frequency calculation. No difference (P < 0.05) was observed between case and control groups using either a weighted or unweighted score (data not shown).
ACCEPTED MANUSCRIPT
4. DISCUSSION
GBS is regarded as an immune-mediated inflammatory demyelinating disease, and a number of previous studies have suggested an association with HLA alleles, and with other candidate genes. However, the involvement of candidate genes in the
development and severity of GBS has remained contentious. To the best of our knowledge, this is the first GWAS studying GBS, and no variants were identified that reached genome-wide significance. What is perhaps most striking from this GWAS, was the lack of any association between MHC SNPS or HLA alleles and GBS.
Recent candidate gene association studies of HLA alleles and GBS have yielded varying results. A study on Iraqi cases and controls found that the frequencies of the HLA genotypes, HLA-DRB4*01:01, HLA-DRB1*03:01 and HLA-DRB1*07:01, were significantly increased in GBS patients [25]. Another study in Tunisians showed an increased prevalence of HLA-DRB1*13 and HLA-DRB1*14 and a decreased
prevalence of HLA-DRB1*03 and HLA-DRB1*07 in GBS patients [26]. A study from Mexico found HLA-DR3 was significantly increased in GBS patients [27]. A recent meta-analysis was unable to find any association with HLA-DQB1 alleles in
Caucasian GBS patients [28], consistent with individual studies finding no HLA Class II associations with GBS [29] When considering different GBS subtypes, two studies of Chinese GBS patients found that epitopes of HLA-DQB1 amino acids, and the allele HLA-DRB1*13, were associated with AIDP compared to AMAN [30, 31]. Furthermore, a study of Indian cases and controls reported that HLA-DRB1*0701 was associated with GBS patients who had preceding infection [32]. In relation to HLA class I genes, Blum et al. [33] performed a study on killer cell immunoglobulin-like receptors (KIRs), molecules involved in the innate immune response, and their HLA ligands. They found that patients with GBS were more likely than controls to have
ACCEPTED MANUSCRIPT
HLA-C2 and HLA-B Bw4 HLA Class I allele groups, which are essential in interactions with KIRs [33].
Although no SNPs with genome-wide or suggestive significance were revealed in the current study, we investigated the most strongly associated SNPs further. The most strongly associated SNP in our study was rs9371600, located in SYNE1 on
chromosome 6. SYNE1 encodes a spectrin repeat-containing protein that localizes to the nuclear membrane. SYNE1 is expressed in multiple tissues, including the central nervous system and skeletal muscle [34]. SYNE1 mutations have been implicated in a number of diseases, including Emery-Dreifuss muscular dystrophy [35],
spinocerebellar ataxia [36] and autosomal recessive arthrogryposis [37], and were observed in a family with two siblings with intellectual disability, spastic paraplegia, axon neuropathy and leukoencephalopathy [38]. As our GWAS cases included all GBS subtypes, including a few cases of MFS in which patients manifest ataxia, it is possible that the signal for rs9371600 was lessened by other GBS subtypes, and further investigation is warranted.
The next most significant SNP, rs16904331, was nominally protective for GBS, and is located in an intergenic region on chromosome 8. The closest gene to this SNP, ADCY8, encodes a membrane-bound adenylate cyclase. ADCY8 is associated with post-traumatic stress disorder, bipolar disorder and alcohol dependence comorbid with depression in females [39]. Chang et al. [40] performed a genome-wide expression analysis of peripheral leukocytes of GBS patients to identify
GBS-associated signaling pathways. The top five SNPs identified in the present study were not among the differentially expressed genes in GBS patients. However, ADCY7 was up-regulated in GBS patients, and featured among six of the ten canonical
pathways identified in the gene-expression analysis [40]. ADCY7 is an adenylate cyclase like ADCY8, the closest gene to the nominally protective variant, rs16904331,
ACCEPTED MANUSCRIPT
identified here. An ADCY7 variant doubles the risk of ulcerative colitis [41], and a possible link between GBS and ulcerative colitis has already been suggested [42, 43], although a causal relationship remains speculative.
Association has previously been reported in a meta-analysis between the TNF-308 SNP and GBS [16]. This association was only observed in comparison of the AA and GG genotypes, only in Asians and not in Caucasians, and not in allelic analyses. On further inspection of the pooled studies, the association is driven by the findings of one Chinese study [44], for which the genotypes overall are not in HWE
(P=4.06x10-12), raising concerns about genotyping accuracy. In the current study we see no association of this variant with GBS, although we cannot exclude association with a variant of small-moderate effect size.
Our study had some limitations. A key issue is the sample size which is too small to identify other than large genetic effects. Although our study with 215 cases is larger than most previous GBS studies, its power was still modest. Under the assumption of a prevalence of approximately 1/10,000, we estimated that we achieved power of 19.6% to detect loci with a MAF = 0.4, additive odds ratio of 1.5, D´ of 1.0 (linkage disequilibrium) and genome-wide significance threshold of P < 5 × 10−8 [45]. Using the same assumptions regarding the genetic model, we estimate that the study had 80% power to detect association with an additive odds ratio of 2.5, and for an additive odds ratio of 2.0 at suggestive levels of association (P < 10−5). As such, a larger cohort achieved from an international collaboration or meta-analysis might be the only way to achieve the required statistical power. Secondly, GBS can be divided into subtypes based on clinical and electrophysiological findings, and the proportions of different subtypes of GBS vary greatly by geography [46]. For the patients with AIDP, the pathology is of inflammation and demyelination, whereas in AMAN there is less inflammation and antibody mediated mechanisms are proposed. For the GBS
ACCEPTED MANUSCRIPT
variants such as MFS and the focal variants, the pathology is also possibly antibody mediated. Thus, multiple mechanisms may lead to GBS, with different immunologic pathways involved among GBS subtypes. In the current study, patients with different GBS subtypes were analysed together. When larger cohorts are available in the future, it will be necessary to conduct analyses on different subtypes.
Our data also suggests that common variant heritability of GBS is low, and therefore that most of the risk of developing GBS is either environmental or due to genetic variants not captured (i.e. directly genotyped or in linkage disequilibrium with genotyped SNPs) using the SNP microarray employed here. So far, no definite prevalence data of GBS have been published, and the incidence of GBS was reported to be 1.1-1.8/100,000 per year [47]. Our study shows that even assuming a prevalence or lifetime risk of GBS as high as 2.5/10,000 or greater the common variant
heritability of the condition remains <0.25. It may be that using denser SNP coverage and including SNPs with lower MAF that a higher heritability would be
demonstrated, but to identify such variants would also require much larger studies. We also cannot exclude the possibility that rare SNPs, copy number variants, or heritable epigenetic variants not tagged by the SNPs studied, contribute to GBS pathogenesis.
GRS analysis can be used for disease risk prediction and to investigate the genetic relatedness between different diseases. In this study, we used GRS to investigate the potential aetiopathogenic relatedness of MS and GBS. Regardless of the algorithm used, whether or not weighted by their effect sizes [48]), GRS based on known MS-associated SNPs did not discriminate between GBS cases and controls. This suggests that although these two diseases are clearly primarily immune mediated and target overlapping components of the nervous system, their aetiopathogeneses are likely to be different.
ACCEPTED MANUSCRIPT
5. CONCLUSIONS
We report the first GWA-based study on GBS. We did not find any risk loci to GBS susceptibility with genome-wide levels of significance or genetic relatedness with another immune-mediated disease with nervous system lesions. This supports the view of previous studies that indicated HLA is not a critical risk factor for GBS pathogenesis. We demonstrate that up to 25% of susceptibility to GBS is determined by common genetic variants. Further larger studies are required, which may also enable the genetic variants associated with specific subsets of GBS to be teased apart.
Acknowledgements
We would like to thank the participating patients and healthy controls for taking part in this study.
Funding: This work was supported by a National Health and Medical Research Council Senior Principal Research Fellowship to MAB.
ACCEPTED MANUSCRIPT
REFERENCES
1. Hughes RA, Cornblath DR. Guillain-Barre syndrome. Lancet. 2005;366(9497):1653-66. doi: 10.1016/S0140-6736(05)67665-9. PubMed PMID: 16271648.
2. Jacobs BC, Rothbarth PH, van der Meche FG, Herbrink P, Schmitz PI, de Klerk MA, et al. The spectrum of antecedent infections in Guillain-Barre syndrome: a case-control study. Neurology. 1998;51(4):1110-5. PubMed PMID: 9781538.
3. Islam Z, Jacobs BC, van Belkum A, Mohammad QD, Islam MB, Herbrink P, et al. Axonal variant of Guillain-Barre syndrome associated with Campylobacter infection in Bangladesh. Neurology. 2010;74(7):581-7. doi: 10.1212/WNL.0b013e3181cff735. PubMed PMID: 20157160.
4. Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky's postulates revisited). Immunol Today. 1993;14(9):426-30. doi: 10.1016/0167-5699(93)90244-F. PubMed PMID: 8216719.
5. Hollenbach JA, Oksenberg JR. The immunogenetics of multiple sclerosis: A comprehensive review. Journal of autoimmunity. 2015;64:13-25. doi: 10.1016/j.jaut.2015.06.010. PubMed PMID: 26142251; PubMed Central PMCID: PMC4687745.
6. Baranzini SE. Revealing the genetic basis of multiple sclerosis: are we there yet? Curr Opin Genet Dev. 2011;21(3):317-24. doi: 10.1016/j.gde.2010.12.006. PubMed PMID: 21247752; PubMed Central PMCID: PMCPMC3105160.
7. Aquil N, Khan IA, Soomro B. Guillain Barre syndrome in a family: a case report of four siblings. Journal of the College of Physicians and Surgeons--Pakistan : JCPSP. 2011;21(3):179-81. doi: 03.2011/JCPSP.179181. PubMed PMID: 21419029.
8. Barzegar M, Rouhi AH, Farhoudi M, Sardashti S. A report of a probable case of familial Guillain Barre syndrome. Annals of Indian Academy of Neurology. 2012;15(4):299-302. doi: 10.4103/0972-2327.104341. PubMed PMID: 23349598;
ACCEPTED MANUSCRIPT
PubMed Central PMCID: PMC3548371.
9. Geleijns K, Brouwer BA, Jacobs BC, Houwing-Duistermaat JJ, van Duijn CM, van Doorn PA. The occurrence of Guillain-Barre syndrome within families. Neurology. 2004;63(9):1747-50. PubMed PMID: 15534275.
10. Naik KR, Saroja AO, Patil BP. Familial Guillain-Barre syndrome: First Indian report. Annals of Indian Academy of Neurology. 2012;15(1):44-7. doi: 10.4103/0972-2327.93278. PubMed PMID: 22412273; PubMed Central PMCID: PMC3299071.
11. Blum S, McCombe PA. Genetics of Guillain-Barre syndrome (GBS) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP): current knowledge and future directions. Journal of the peripheral nervous system : JPNS. 2014;19(2):88-103. doi: 10.1111/jns5.12074. PubMed PMID: 25039604.
12. Seldin MF. The genetics of human autoimmune disease: A perspective on progress in the field and future directions. Journal of autoimmunity. 2015;64:1-12. doi: 10.1016/j.jaut.2015.08.015. PubMed PMID: 26343334; PubMed Central PMCID: PMCPMC4628839.
13. Stewart GJ, Pollard JD, McLeod JG, Wolnizer CM. HLA antigens in the Landry-Guillain-Barre syndrome and chronic relapsing polyneuritis. Annals of neurology. 1978;4(3):285-9. doi: 10.1002/ana.410040317. PubMed PMID: 718142. 14. Latovitzki N, Suciu-Foca N, Penn AS, Olarte MR, Chutorian AM. HLA typing and Guillain-Barre syndrome. Neurology. 1979;29(5):743-5. PubMed PMID: 571573. 15. Winer JB, Briggs D, Welsh K, Hughes RA. HLA antigens in the Guillain-Barre syndrome. Journal of neuroimmunology. 1988;18(1):13-6. PubMed PMID: 2831249. 16. Liu J, Lian Z, Chen H, Shi Z, Feng H, Du Q, et al. Associations between tumour necrosis factor-alpha gene polymorphisms and the risk of Guillain-Barre syndrome and its subtypes: A systematic review and meta-analysis. Journal of neuroimmunology. 2017;313:25-55.
ACCEPTED MANUSCRIPT
Guillain-Barre syndrome. Annals of neurology. 1990;27 Suppl:S21-4. PubMed PMID: 2194422.
18. Sejvar JJ, Kohl KS, Gidudu J, Amato A, Bakshi N, Baxter R, et al. Guillain-Barre syndrome and Fisher syndrome: case definitions and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine. 2011;29(3):599-612. doi: 10.1016/j.vaccine.2010.06.003. PubMed PMID: 20600491.
19. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. PubMed PMID: 25722852; PubMed Central PMCID: PMC4342193.
20. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics. 2007;81(3):559-75. doi: 10.1086/519795. PubMed PMID: 17701901; PubMed Central PMCID: PMC1950838.
21. Jia X, Han B, Onengut-Gumuscu S, Chen WM, Concannon PJ, Rich SS, et al. Imputing amino acid polymorphisms in human leukocyte antigens. PloS one. 2013;8(6):e64683. doi: 10.1371/journal.pone.0064683. PubMed PMID: 23762245; PubMed Central PMCID: PMC3675122.
22. Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. American journal of human genetics. 2011;88(1):76-82. doi: 10.1016/j.ajhg.2010.11.011. PubMed PMID: 21167468; PubMed Central PMCID: PMC3014363.
23. Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. American journal of human genetics. 2011;88(3):294-305. doi: 10.1016/j.ajhg.2011.02.002. PubMed PMID: 21376301; PubMed Central PMCID: PMC3059431.
ACCEPTED MANUSCRIPT
NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(Database issue):D1001-6. doi: 10.1093/nar/gkt1229. PubMed PMID: 24316577; PubMed Central PMCID: PMCPMC3965119.
25. Hasan ZN, Zalzala HH, Mohammedsalih HR, Mahdi BM, Abid LA, Shakir ZN, et al. Association between human leukocyte antigen-DR and demylinating Guillain-Barre syndrome. Neurosciences. 2014;19(4):301-5. PubMed PMID: 25274590; PubMed Central PMCID: PMC4727669.
26. Fekih-Mrissa N, Mrad M, Riahi A, Sayeh A, Zaouali J, Gritli N, et al. Association of HLA-DR/DQ polymorphisms with Guillain-Barre syndrome in Tunisian patients. Clinical neurology and neurosurgery. 2014;121:19-22. doi: 10.1016/j.clineuro.2014.03.014. PubMed PMID: 24793468.
27. Gorodezky C, Varela B, Castro-Escobar LE, Chavez-Negrete A, Escobar-Gutiérrez A, Martinez-Mata J. HLA-DR antigens in Mexican patients with Guillain-Barré syndrome. Journal of neuroimmunology. 1983;4(1):1-7. PubMed PMID: 6401765.
28. Jin PP, Sun LL, Ding BJ, Qin N, Zhou B, Xia F, et al. Human Leukocyte Antigen DQB1 (HLA-DQB1) Polymorphisms and the Risk for Guillain-Barre Syndrome: A Systematic Review and Meta-Analysis. PloS one. 2015;10(7):e0131374. doi: 10.1371/journal.pone.0131374. PubMed PMID: 26204120; PubMed Central PMCID: PMC4512729.
29. Geleijns K, Schreuder GM, Jacobs BC, Sintnicolaas K, van Koningsveld R, Meulstee J, et al. HLA class II alleles are not a general susceptibility factor in Guillain-Barre syndrome. Neurology. 2005;64(1):44-9. doi: 10.1212/01.WNL.0000148727.02732.01. PubMed PMID: 15642902.
30. Magira EE, Papaioakim M, Nachamkin I, Asbury AK, Li CY, Ho TW, et al. Differential distribution of HLA-DQ beta/DR beta epitopes in the two forms of Guillain-Barre syndrome, acute motor axonal neuropathy and acute inflammatory demyelinating polyneuropathy (AIDP): identification of DQ beta epitopes associated
ACCEPTED MANUSCRIPT
with susceptibility to and protection from AIDP. Journal of immunology. 2003;170(6):3074-80. PubMed PMID: 12626563.
31. Monos DS, Papaioakim M, Ho TW, Li CY, McKhann GM. Differential distribution of HLA alleles in two forms of Guillain-Barre syndrome. The Journal of infectious diseases. 1997;176 Suppl 2:S180-2. PubMed PMID: 9396707.
32. Sinha S, Prasad KN, Jain D, Nyati KK, Pradhan S, Agrawal S. Immunoglobulin IgG Fc-receptor polymorphisms and HLA class II molecules in Guillain-Barre syndrome. Acta neurologica Scandinavica. 2010;122(1):21-6. doi: 10.1111/j.1600-0404.2009.01229.x. PubMed PMID: 20105138.
33. Blum S, Csurhes P, Reddel S, Spies J, McCombe P. Killer immunoglobulin-like receptor and their HLA ligands in Guillain-Barre Syndrome. Journal of neuroimmunology. 2014;267(1-2):92-6. doi: 10.1016/j.jneuroim.2013.12.007. PubMed PMID: 24367901.
34. Fanin M, Savarese M, Nascimbeni AC, Di Fruscio G, Pastorello E, Tasca E, et al. Dominant muscular dystrophy with a novel SYNE1 gene mutation. Muscle & nerve. 2015;51(1):145-7. doi: 10.1002/mus.24357. PubMed PMID: 25091525.
35. Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A, et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Human molecular genetics. 2007;16(23):2816-33. doi: 10.1093/hmg/ddm238. PubMed PMID: 17761684.
36. Gros-Louis F, Dupre N, Dion P, Fox MA, Laurent S, Verreault S, et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nature genetics. 2007;39(1):80-5. doi: 10.1038/ng1927. PubMed PMID: 17159980. 37. Attali R, Warwar N, Israel A, Gurt I, McNally E, Puckelwartz M, et al. Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Human molecular genetics. 2009;18(18):3462-9. doi: 10.1093/hmg/ddp290. PubMed PMID: 19542096.
ACCEPTED MANUSCRIPT
Bon BW, de Ligt J, et al. Identification of pathogenic gene variants in small families with intellectually disabled siblings by exome sequencing. Journal of medical genetics. 2013;50(12):802-11. doi: 10.1136/jmedgenet-2013-101644. PubMed PMID: 24123876.
39. Wolf EJ, Rasmusson AM, Mitchell KS, Logue MW, Baldwin CT, Miller MW. A genome-wide association study of clinical symptoms of dissociation in a trauma-exposed sample. Depression and anxiety. 2014;31(4):352-60. doi: 10.1002/da.22260. PubMed PMID: 24677629; PubMed Central PMCID: PMC3984628.
40. Chang KH, Chuang TJ, Lyu RK, Ro LS, Wu YR, Chang HS, et al. Identification of gene networks and pathways associated with Guillain-Barre syndrome. PloS one. 2012;7(1):e29506. doi: 10.1371/journal.pone.0029506. PubMed PMID: 22253732; PubMed Central PMCID: PMCPMC3254618.
41. Luo Y, de Lange KM, Jostins L, Moutsianas L, Randall J, Kennedy NA, et al. Exploring the genetic architecture of inflammatory bowel disease by whole-genome sequencing identifies association at ADCY7. Nature genetics. 2017;49(2):186-92. doi: 10.1038/ng.3761. PubMed PMID: 28067910; PubMed Central PMCID: PMCPMC5289625.
42. Krystallis CS, Kamberoglou DK, Cheilakos GB, Maltezou MN, Tzias VD. Guillain-Barre syndrome during a relapse of ulcerative colitis: a case report. Inflamm Bowel Dis. 2010;16(4):555-6. doi: 10.1002/ibd.21071. PubMed PMID: 19714764. 43. Zimmerman J, Steiner I, Gavish D, Argov Z. Guillain-Barre syndrome: a possible extraintestinal manifestation of ulcerative colitis. J Clin Gastroenterol. 1985;7(4):301-3. PubMed PMID: 4045173.
44. Jiao H, Wang W, Wang H, Wu Y, Wang L. Tumor necrosis factor alpha 308 G/A polymorphism and Guillain-Barre syndrome risk. Mol Biol Rep. 2012;39(2):1537-40. doi: 10.1007/s11033-011-0892-1. PubMed PMID: 21604171.
ACCEPTED MANUSCRIPT
association genetic mapping studies of complex traits. Bioinformatics. 2003;19(1):149-50. PubMed PMID: 12499305.
46. Kuwabara S, Yuki N. Axonal Guillain-Barre syndrome: concepts and controversies. The Lancet Neurology. 2013;12(12):1180-8. doi: 10.1016/S1474-4422(13)70215-1. PubMed PMID: 24229616.
47. McGrogan A, Madle GC, Seaman HE, de Vries CS. The epidemiology of Guillain-Barre syndrome worldwide. A systematic literature review. Neuroepidemiology. 2009;32(2):150-63. doi: 10.1159/000184748. PubMed PMID: 19088488.
48. De Jager PL, Chibnik LB, Cui J, Reischl J, Lehr S, Simon KC, et al. Integration of genetic risk factors into a clinical algorithm for multiple sclerosis susceptibility: a weighted genetic risk score. The Lancet Neurology. 2009;8(12):1111-9. doi: 10.1016/S1474-4422(09)70275-3. PubMed PMID: 19879194; PubMed Central PMCID: PMC3099419.
ACCEPTED MANUSCRIPT
Table 1. Patient characteristics.
Patient Characteristica Number
Gender Male Female 135 80 Brighton Criteria Diagnostic Certainty Level 1 Level 2 Level 3 Level 4 68 107 20 20 HughesGBS Disability Scaleb 1 2 3 4 5 6 2 22 35 89 43 0 GBS Subtype AIDP GBS, unspecified AMAN MFS Other subtypes 105 73 8 17 12
aThe median age of onset was 55.4 years of age, range 15.4–89.1 (data was available for 212 of the 215 patients).
ACCEPTED MANUSCRIPT
Table 2. Detailed information for quality control filtering.
Individuals or Markers remaining after each QC-filtering step (n)
Cases Controls Markers
Before QC 215 1,105 699,609
MIND < 0.1 215 1,102 −
GENO < 0.05 − − 695,656
HWE > 10−6 − − 693,288
MAF > 0.05 − − 581,413
Different missing rate between groups − − 579,499 IBD < 0.185 204 798 − Heterozygosity (mean±3SD) with MIND < 0.1 199 756 − PCA 191 639 −
Final data for association analysis
191 639 579,499
GENO, missing rate per SNP; HWE, Hardy-Weinberg equilibrium; IBD, identity by descent; MAF, minor allele frequency; MIND, missingness per individual; n, number; PCA, principal component analysis; QC, quality control; SD, standard deviation.
ACCEPTED MANUSCRIPT
Table 3. Detailed information of the five most significant SNPs.
Chr, chromosome; OR, odds ratio.
SNP Chr Position Locus/closest gene Minor allele OR P-value rs9371600 6 152790533 SYNE1 A 1.83 1.56 × 10−5 rs16904331 8 131651760 ADCY8 G 0.33 1.65 × 10−5 rs2243523 17 80680449 FN3KRP T 1.70 1.91 × 10−5 rs7733891 5 75099610 POC5 G 2.66 1.96 × 10−5 rs10506873 12 82792942 METTL25 G 2.23 1.99 × 10−5
ACCEPTED MANUSCRIPT
FIGURE LEGENDS
Figure 1. Manhattan plot of SNP associations with GBS. Blue line, P = 10−5; red line, P = 5 × 10−8.
Figure 2. Estimated common-variant SNP heritability of GBS in relation to disease prevalence, with 95% confidence intervals indicated by vertical bars.
ACCEPTED MANUSCRIPT
HIGHLIGHTS
Guillain-Barré syndrome (GBS) is considered to have an autoimmune basis.
A genetic contribution to GBS has been controversial, thus we performed a GWAS.
No single genetic variant (neither tagged nor imputed SNP) was associated with GBS
Common genetic variants contribute up to 25% of susceptibility to GBS.