Pituitary Tumor Registry: A Novel Clinical Resource*
MARLYS R. DRANGE, NICOLE R. FRAM, VIVIEN HERMAN-BONERT, AND
SHLOMO MELMED
Cedars-Sinai Research Institute, University of California–Los Angeles School of Medicine, Los Angeles, California 90048
ABSTRACT
Pituitary adenomas result in clinical sequelae and accelerated mortality due to central mass effects or pituitary hormone hyperse-cretion and/or insufficiency. The low annual incidence and prolonged natural history of these rare tumors has hindered efforts to evaluate long-term clinical outcomes. Care of these patients is often provided by larger tertiary specialist referral centers. A novel evidence-based computerized pituitary tumor registry was developed to systemati-cally evaluate epidemiological, biochemical, and clinical outcome data. Retrospective registration of 371 patients [99 clinically non-functioning tumors (CNFTs), 176 acromegalics, and 96 prolactino-mas] with radiological, biochemical, and clinical evidence of pituitary tumors was performed. Analysis of this primarily specialist-referred population revealed a female predominance among CNFT (60%) and prolactinoma (69%) patients. Males had a significantly greater
fre-quency of macroadenomas than females for CNFTs (92%vs.(68%) and
for prolactinomas (74%vs.40%). Males with prolactinomas also had
higher mean pretreatment serum PRL levels (1206vs.219 ng/mL).
Concurrent hyperprolactinemia was present in CNFT (47%) and ac-romegaly (33%) patients. Radiographic cure, defined as absence of visualized tumor, was achieved in 21% of CNFTs, 34% of acromeg-alies, and 21% of prolactinomas. Biochemical remission, defined by normalization of hormonal tumor markers, was observed in 35% of acromegaly and 39% of prolactinoma patients in the registry, thus reflecting the tertiary referral patterns. Nine premature deaths
(pa-tients aged #65 yr) occurred in the acromegaly subpopulation,
whereas no premature deaths were encountered in nonacromegalic patients.
In conclusion, this unique and comprehensive pituitary tumor reg-istry enables identification of diagnostic and prognostic markers and evaluation of long-term clinical outcomes. Prospectively, this registry will improve therapeutic guidelines and cost-effective pituitary tumor
management. (J Clin Endocrinol Metab85:168 –174, 2000)
S
ILENT PITUITARY microadenomas are identified in ap-proximately 11% of autopsy specimens (1). However, symptomatic pituitary tumors are rarely encountered clini-cally. Based on a study of the Olmstead County population from 1970 –1980, the estimated mean annual incidence of pituitary tumors ranges from 0.5 to 7.4 per 100,000 persons depending on age and sex (2), with the highest incidence occurring in women aged 15– 44 y. Tumors of pituitary origin are usually slow-growing, benign, monoclonal neoplasms, yet are associated with significant morbidity and premature mortality. Pituitary adenomas come to clinical attention due to mass effects resulting from impingement on local struc-tures or manifestations of pituitary hormone excess or in-sufficiency (3–7).These tumors are classified according to their cell type of origin. Each cell type produces specific hormone products under tightly regulated hypothalamic and peripheral con-trol. The hormone-secreting cells may give rise to functional pituitary tumors that hypersecrete one or more hormones including PRL, GH, ACTH, and, rarely, FSH, LH, or TSH. Pituitary tumors with no identifiable clinical hypersecretory syndrome are termed clinically nonfunctioning tumors (CNFTs) because the majority of these tumors are, in fact, of
gonadotroph cell origin and actually express, albeit ineffi-ciently, FSH, LH, or their free subunits (6).
Compared with other neoplasms, pituitary adenomas tend to occur in younger patients. Successful management aimed at normalization of hormonal excess or deficiency, decom-pression of vital structures, and prevention of tumor recur-rence often requires lifelong treatment and follow-up. The relatively low annual incidence of pituitary adenoma diag-nosis and long natural history of these tumors have ham-pered efforts to evaluate remote clinical outcomes in signif-icant numbers of patients. Moreover, the various tumor types must be evaluated separately because each has a unique frequency, pathology, therapeutic regimen, and long-term outcome. Reliable tumor type-specific epidemiological in-formation is sparse or nonexistent.
We, therefore, developed a novel evidence-based com-puterized pituitary tumor registry to collect comprehen-sive demographic, therapeutic, and clinical outcome in-formation on patients harboring pituitary mass lesions of all types. Our clinic is primarily a tertiary resource for specialist referrals. This data acquisition and evaluation tool provides epidemiological, biochemical, and outcome data for pituitary tumor patients to define features of prognostic significance. The goal of the Pituitary Tumor Registry is improvement of early identification of these patients, tumor management strategies, and long-term outcome. To illustrate the utility of this unique tumor registry, we describe a number of retrospective analyses for the three most common pituitary tumor types: CNFT, acromegaly, and prolactinoma.
Received August 9, 1999. Revision received October 5, 1999. Accepted October 11, 1999.
Address correspondence and requests for reprints to: Shlomo Melmed, Cedars-Sinai Medical Center, 8700 Beverly Boulevard, Room 2015, Los Angeles, California 90048. E-mail: Melmed@cshs.org.
* Supported by NIH Grant CA-75979; Lilly Research Laboratories, The Steinberg Foundation and a grant from the Endocrine Fellows Foundation (to M.R.D.).
Copyright © 2000 by The Endocrine Society
Subjects and Methods Human subjects
Patient participation was approved by the Cedars-Sinai Medical Cen-ter and Los Angeles County/University of Southern California Institu-tional Review Boards.
Patient population
Hospital chart review was performed by two physicians who also directly supervised data entry by a data analyst of tertiary referral patients seen at the Cedars-Sinai Medical Center and at the Los Angeles County Hospital from 1982–1999. When appropriate, physicians or pa-tients were contacted for information. To date, 404 papa-tients [99 CNFT, 176 acromegaly (GH-secreting), 96 prolactinoma, 18 Cushing’s (ACTH-secreting), and 15 other nonpituitary intrasellar tumors) have been reg-istered. Further analysis of patients bearing Cushing’s and other intra-sellar masses was not undertaken due to the paucity of these tumor types in the registry. Patients included in the pituitary tumor registry had clinical, biochemical, and radiological evidence of an intrasellar mass lesion. All types and classes of tumors were included, and there were no exclusion criteria. Tumor type was determined by hormonal biochem-ical profile and clinbiochem-ical presentation. Immunohistochembiochem-ical confirma-tion was not always available, either because the patient had not un-dergone surgical excision of the tumor, immunohistochemistry had not been performed at the time of pathological evaluation of the tumor tissue, or pathology reports were not available. Ethnic distribution of registered patients was 69% Caucasian, 19% Hispanic, 8% Asian, and 4% African American. Males accounted for 41% and females accounted for 59% of patients. Patient ages ranged from 8 – 89 y.
Pituitary tumor registry computer program
The computer software Access 97 (Microsoft Corporation, 1996) was used for development of the registry program. Selection of specific data for registry inclusion was based on review of the current literature regarding prognostic factors for aggressive pituitary tumors and unfa-vorable clinical outcomes. Factors known to have prognostic signifi-cance, those with potential signifisignifi-cance, but with insufficient confirma-tory information, and possible confounding factors were also included. Data collection worksheets that paralleled the computer program and flow sheets for tracking laboratory results or medication changes were designed using Powerpoint 97 (Microsoft Corporation, 1996). These worksheets were developed to achieve maximum data ascertainment, standardized responses, and to create an efficient, user-friendly system for detailed data collection. To maintain patient confidentiality, data were entered using an arbitrarily assigned identification number. Only individuals registered as principal or coinvestigators had access to iden-tifying information.
Definitions
Tumor size was determined by radiographic analysis. Tumors less than 10 mm in greatest dimension were considered microadenomas and tumors greater than or equal to 10 mm were considered macroadeno-mas. Hypogonadal symptoms included complaints of diminished li-bido, oligo- or amenorrhea, infertility, or erectile dysfunction. All lab-oratory values were interpreted based on the reporting lablab-oratory’s normal range, unless stated otherwise. Concurrent hyperprolactinemia was defined as PRL elevation (upper limits of normal ranged from 14.7–30mg/L depending on laboratory) in a patient diagnosed with either CNFT or acromegaly. Hypogonadotropic hypogonadism was diagnosed when estradiol or testosterone levels were low in the presence of inappropriately normal or low FSH and/or LH levels. Hypopituitar-ism included deficiency of the GH (evoked GH,3 ng/mL), gonadal (FSH/LH), thyroid (abnormal thyroid hormone levels with inappropri-ate TSH levels), adrenal (blunted cortrosyn responses) axes, and/or panhypopituitarism. Radiographic cure was defined as the absence of visualized tumor tissue on magnetic resonance (MRI), sensitivity more than 2 mm, or computed tomography imaging (if MRI scans were not available). Biochemical control of prolactinoma was defined as normal-ized serum PRL levels. Biochemical remission of acromegaly was de-fined by the strict criteria of both a normalized insulin-like growth
factor-I (IGF-I) for age and sex and a glucose- (or somatostatin analog-) suppressed GH value less than or equal to 2mg/L.
Statistical analysis
Patient groups were generally compared by Fisher’s two-tailed exact test. Gender predominance was determined by comparison to an ex-pected rate of 50% using Yates correctedx2analysis. Because the dis-tribution of pretreatment PRL levels was skewed, the data were log transformed before application of the two-samplettest and the geo-metric means are reported. Standardized mortality ratio (SMR) was computed by standard methods as described previously (7). Diagnosis date and death date or date of last contact were entered as the dates of study entry and exit, respectively. Death rates were obtained for males and females using the “all races” rates in the National Vital Statistics Report [1998, 47(13):11].
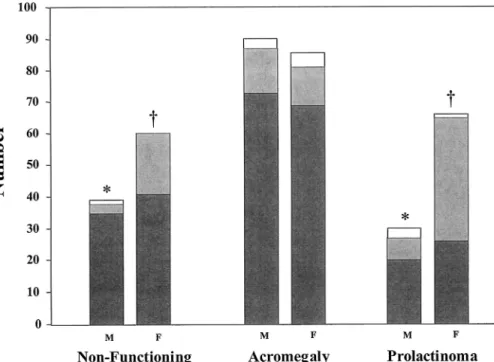
Results Patient gender and tumor size (Fig. 1)
From 1982 through current (1999), nearly three-quarters of all patients registered harbored macroadenomas (76 of 98 CNFTs, 142 of 168 acromegalies, and 46 of 92 prolactinomas). CNFTs (60%, n599,P,0.05) and prolactinomas (69%, n5 96,P,0.01) were more frequent in women in contrast to the equal gender distribution for patients with acromegaly (51% male, 49% female, n5176) in the registry. Males with pro-lactinomas tended to have larger tumors than females with this tumor type, 74% (n527) macroadenomasvs.40% (n5 65, P , 0.01), and higher mean pretreatment serum PRL levels, 1206 (n522)vs.219 ng/mL (n543,P,0.001). Males with CNFTs also had a significantly greater (P,0.01) like-lihood to have a macroadenoma, 92% (n538) compared to 68% (n 560) for females. Distribution of tumor size was equal between men and women with GH-secreting tumors (84% and 85% macroadenomas, respectively).
Diagnostic information and symptoms
The age of patients at the time of diagnosis peaked in the third decade for acromegaly, the fourth decade for prolacti-nomas, and across the fourth and fifth decades for CNFT (Fig. 2). The mean time delay from reported symptom onset to diagnosis was 3.063.9 y for prolactinomas (n584) and 1.96 2.9 y for CNFTs (n577) compared to 7.267.7 y for acro-megalies (n5146). Internists or family physicians (29%) and endocrinologists (24%) were primarily responsible for mak-ing the initial diagnosis of pituitary lesions (n5261). Tumor type-specific distribution of the diagnostic source is illus-trated in Table 1. Differences in diagnostic source correlates with the particular symptomatology associated with each tumor type that is listed in Table 2. Patients with CNFTs generally presented with mass-related effects such as head-ache (48%) and visual deficits (48%). Those with acromegaly most frequently reported features unique to GH excess, such as acral enlargement (86%) and maxillofacial changes (74%). Patients with PRL-secreting tumors presented most com-monly with hypogonadal symptoms (70%), galactorrhea (49%), and headache (46%). Since galactorrhea is uncommon in men (8) and only 4% (3/82) of patients in the registry reporting this symptom were male, the incidence of galac-torrhea was analyzed separately for female patients. Thirty-two percent (n559) of female patients harboring CNFTs, 17% (n586) with acromegaly and 68% (n566) with
pro-lactinomas, reported galactorrhea as a presenting clinical feature. Not surprisingly, patients complaining of headache more commonly had macroadenomas for GH-secreting tu-mors (93%, n567) and CNFTs (81%, n548). In contrast, reports of headache by patients with PRL-secreting tumors
(n541) were more evenly distributed between small (44%) and large (56%) tumors, suggesting that factors in addition to tumor size-related mass effect may contribute to incidence of headache in patients with prolactinomas. Hypogonadal symptoms were more commonly observed in patients with CNFT microadenomas (34%, n535) than GH-secreting tu-mors of similar size (11%, n565).
Concurrent hormonal aberrations (Table 3)
Concurrent hyperprolactinemia was present in 47% (n5 73) of CNFT and 33% (n 5 140) of acromegaly patients. Thirty-two percent of CNFTs with hyperprolactinemia were less than 1 cm in size compared to 5% of acromegaly tumors exhibiting concurrent hyperprolactinemia. Partial or com-plete pituitary hormone insufficiency was present at diag-nosis in 44% of CNFT (n566), 38% of acromegaly (n5130), and 71% of prolactinoma patients (n580). The gonadal axis (FSH/LH) was affected in nearly all cases (93%, 96%, and
FIG. 1. Distribution of tumor type by
size ( , microadenoma; f,
macroad-enoma; u, tumor size unknown) and
gender. Asterisk (*) indicates statistical signficance of tumor size, and dagger (†) indicates significance of gender.
FIG. 2. Age at diagnosis in patients with clinically nonfunctioning
pituitary tumors (m, n597), acromegaly (v, n5175), and
prolacti-nomas (Œ, n595).
TABLE 1. Distribution of initial diagnostic source by tumor type:
clinically nonfunctioning (n571), acromegaly (n5126), and
prolactinoma (n564) Diagnostic source Total percentage Nonfunc-tioning Acro-megaly Prolac-tinoma Internist/family medicine 23 39 16 Endocrinologist 14 25 34 Gynecologist 10 ,1 34 Ophthalmologist/optometrist 25 6 6 Relatives/self 1 6 2 Neurologist 3 5 0 Dentist 0 5 0 Other 24 14 8
TABLE 2. Tumor-specific presenting clinical features of pituitary/ sellar tumors Feature Total percentage Nonfunctioning (n599) Acromegaly (n5176) Prolactinoma (n596) Hypogonadal symptoms 35 38 70 Headache 48 40 46 Galactorrhea 19 9 49 Visual deficit 48 26 19 Acral enlargement — 86 — Maxillofacial changes — 74 — Excessive perspiration — 48 — Arthralgia — 46 — Weight gain 13 18 13 Fatigue 20 26 17
The most frequent features observed for each tumor type are shown
inboldprint. Hypogonadal symptoms include complaints of decreased
100%, respectively). Concurrent pretreatment hypogonado-tropic hypogonadism was also more likely to occur in pa-tients bearing CNFT microadenomas (26%, n 5 27) than GH-secreting microadenomas (4%, n546). Indeed, the in-cidence of hypogonadotropic hypogonadism paralleled that of concurrent hyperprolactinemia in these patients. Hypop-ituitarism occurred as a treatment complication in 32% (n5 78) of CNFT, 34% (n5140) of acromegaly, and only 13% (n5 86) of prolactinoma patients. Overall, partial or complete pituitary insufficiency was encountered in 65% of CNFT (n5 66), 63% of acromegaly (n5124) and 49% of prolactinoma (n 5 76) patients following treatment. Persistent diabetes insipidus was present in 14% of CNFT (n 5 66), 10% of acromegaly (n 5 124) and 3% of prolactinoma (n 5 76) patients. Hypopituitarism and diabetes insipidus were con-sistently associated with large tumors (80%, n 5159,P,
0.001).
Therapy and clinical outcome
Several treatment modes, including surgery, irradiation, and pharmacotherapy, are currently available for the man-agement of pituitary tumors. Utilization patterns of these individually and as combinations for each tumor type are listed in Table 4. Surgery alone was the most common mo-dality used in the treatment of CNFT (46%, n 5 99), and medical treatment with dopamine agonists (either bro-mocriptine or the newer long-acting agent cabergoline) was the mainstay of prolactinoma therapy (53%, n596). In this tertiary patient population, a combination of surgical and medical therapy (40%, n5176) or all modalities (21%) was used for the management of GH-secreting tumors. Pharma-cotherapy with either somatostatin analogs (39%, n 590), such as octreotide, dopamine agonists (16%), or both (46%) was used as an adjunctive therapy after surgery. Several of the registered patients were also enrolled in clinical trials that used novel sustained-release formulations of somatostatin analogs or GH receptor antagonists.
Radiographic cure was achieved in 21% of CNFTs (n558), 34% of GH-secreting tumors (n5137), and 21% of prolacti-nomas (n 5 72). Biochemical remission, by the stringent criteria defined, was achieved in 35% of ascertainable acro-megaly patients (n 5124), whereas biochemical remission
resulting in sustained normalization of serum PRL levels was achieved in 39% of prolactinoma patients (n594). Further evaluation of the characteristics of cured patients revealed that the majority of biochemically controlled prolactinomas, 62% (n537), were less than 1 cm in size. However, 79% (n5 43) of biochemically controlled acromegaly patients and 70% of radiographically-cured tumors (10 of 12 CNFTs, 34 of 44 acromegalies, and 4 of 13 prolactinomas) were macroadenomas.
Nine deaths occurred in acromegaly patients aged 40 – 65 y. These deaths were caused by cardiovascular/cerebrovas-cular disease, malignancy, respiratory disease, diabetic com-plications, and suicide (three, two, two, one, and one patient, respectively). No premature deaths were identified for the other tumor types. The computed SMR is 0.85 (confidence interval, 0.42–1.52).
Discussion
The goals of this project are to improve early identification of patients with pituitary tumors, define management strat-egies, and provide a basis for assessing long-term outcomes. Tumor registries are an important resource for the study of cancer epidemiology, treatment strategies, and long-term clinical outcomes. Thus, we developed a comprehensive computerized tumor registry program for data collection on patients with pituitary/intrasellar mass lesions. Using this prototype database program, retrospective demographic,
TABLE 3. Frequency of associated hormonal abnormalities observed pretreatment, following therapeutic intervention, and occurring as a complication of therapy for each tumor type
Total percentage
Nonfunctioning Acromegaly Prolactinoma
Pretreatment (n566) (n5130) (n580) Concurrent hyperprolactinemia 47 33 NA Hypopituitarism 44 38 71 Macroadenoma 74 96 53 Posttreatment (n566) (n5124) (n576) Hypopituitarism 65 63 49 Macroadenoma 83 87 64 Diabetes insipidus 14 10 3 Therapeutic complications (n578) (n5140) (n586) Hypopituitarism 32 34 13 Diabetes insipidus-transient 21 8 9 Diabetes insipidus-persistent 12 7 2
Hypopituitarism refers to insufficiency of one or more anterior pituitary hormonal axes. NA, Not applicable.
TABLE 4. Therapeutic modalities used for each tumor type:
nonfunctioning (n599), acromegaly (n5176), and prolactinoma
(n596).
Therapy Total percentage
Nonfunctioning Acromegaly Prolactinoma
Surgery alone 46 18 5 Medical alone 13 12 53 Irradiation alone 2 ,1 0 Surgery1medical 9 40 35 Surgery1irradiation 12 3 1 All modalities 1 21 2 Observation 16 2 3
The most common therapy for each tumor type is shown inbold
clinical, and biochemical presentation, therapeutic and out-come data for the three most common pituitary tumor types—CNFTs, GH-secreting tumors, and prolactinomas— were analyzed. Patients encountered in this study were largely tertiary referrals from other specialists. Thus, the results depict these parameters in a select group of patients who may have more complex disease and do not necessarily reflect characteristics of the overall population.
Results for prolactinomas revealed a female predomi-nance, although male patients had a greater frequency of macroadenomas and higher pretreatment serum PRL levels. The higher hormone levels observed in male patients are most likely a direct reflection of tumor size, as these param-eters are generally concordant (9). These findings are con-sistent with previous studies (10, 11) and in contrast to post-mortem studies that indicated an equal distribution of clinically nonsignificant microprolactinomas encountered across adult age groups and between sexes (8). Estrogen is known to stimulate lactotroph proliferation and PRL gene transcription leading to increased PRL synthesis (12, 13). Taken together, these results suggest that the female sex, perhaps due to estrogen, is associated with tumor promotion, but additional nongender related factors are required to con-fer tumor aggressiveness or invasiveness.
CNFT diagnosis was also significantly more frequent in women, with men more commonly bearing macroadenomas than their female counterparts. These results are similar to prolactinomas and in contrast to GH-secreting tumors in which both gender and macroadenomas were equally dis-tributed. These observations may reflect gender differences in patient inclination to seek medical attention for hypogo-nadal symptoms. Conversely, this may reflect a true as yet unexplored gender-based difference in the biologic pheno-type of CNFT similar to that observed for prolactinoma.
Analysis of these results demonstrated that clinical onset and diagnosis of the majority of pituitary tumors occurred in young adults. More patients bearing CNFT, as compared to acromegaly and prolactinoma, were diagnosed past 40 yr of age presumably due to the absence of overt clinical mani-festations until this slow-growing tumor caused pressure-related effects on local central structures. These data also confirm previous findings that the clinical effects of GH excess present insidiously, such that there was an average of 7.2 y delay between reported time of symptom onset and diagnosis of acromegaly. This figure is comparable to earlier studies that report delays on the order of 7 yr (14, 15) and likely underestimates the actual time lapse. Importantly, these results should challenge the practicing clinician to en-tertain the diagnosis of acromegaly earlier in the course of this debilitating disease.
Hyperprolactinemia was observed in one third of patients with acromegaly. This finding was not unexpected because GH-secreting (somatotrophs) and PRL-secreting cells (lac-totrophs) derive from a common progenitor cell, the mam-mosomatotroph (16). Moreover, the frequency observed for our patient population is similar to previous reports that indicate approximately 35% of acromegalic patients have concomitant hypersecretion of PRL and GH (17). Alterna-tively, moderate serum PRL elevation may be due to “stalk section effect” caused by disruption of dopaminergic
inhi-bition of PRL synthesis and secretion. We also observed concurrent PRL elevation in 47% of CNFTs. This frequency is similar to that reported (42– 65%) in the few studies ad-dressing this phenomenon (summarized in Ref. 18). Serum PRL elevation in these patients was mild, usually less than 100mg/L, suggesting that the stalk-section phenomenon is the underlying etiology for concurrent hyperprolactinemia observed in CNFT patients. Stalk-section effect is not specific for large tumors. Smith and Laws (19) found no significant correlation between PRL levels and tumor size, stalk devi-ation, or degree of stalk compression as assessed by MRI in a series of 44 pathologically confirmed PRL-negative tumors. Indeed, in the results herein, 32% of the CNFTs associated with secondary hyperprolactinemia were microadenomas. Conversely, raised intrasellar pituitary tissue pressure has been associated with hypopituitarism and hyperprolactine-mia and may explain these findings (20, 21). Alternatively, some small prolactinomas producing proportionately low PRL levels may have been misclassified as CNFTs. The only definitive way to confirm tumor type is to demonstrate tu-mor cell PRL productionin vitroby PRL immunohistochem-ical staining. These data demonstrating mild to moderate elevations in serum PRL in CNFTs and acromegaly illustrate the need to routinely obtain an MRI and include a screening IGF-I level to rule out acromegaly, CNFT, and nonpituitary sellar or hypothalamic masses in the evaluation of low-level hyperprolactinemia.
Pituitary insufficiency can be encountered in patients with any size or type of intrasellar mass and, thus, serves as an important diagnostic indicator of a pituitary lesion (3). Hy-pogonadotropic hypogonadism was a relatively common pretherapeutic finding for all tumor types and corresponds to the frequency of hypogonadal symptoms at presentation. The frequency of hormone deficiency loss generally follows the order GH.FSH/LH.TSH.ACTH (3). These results suggest that an even greater percentage of patients may have been GH deficient. Adequacy of the GH axis was rarely evaluated in the past, but remains to be explored as an additional tumor surveillance marker.
A higher prevalence of hypogonadotrophic hypogonad-ism was observed in CNFT patients than in those with ac-romegaly. This difference does not appear to be tumor size-dependent, suggesting that the mechanism is not tumoral destruction of the adjacent gonadotrophs by direct invasion or compression. Similar findings were previously reported by Greenmanet al.(22) and Damjanovicet al.(23). Interest-ingly, we found that the presence of hypogonadism in these patients paralleled that of concurrent hyperprolactinemia. Indeed, hyperprolactinemia may explain the presence of FSH/LH deficiency in these patients because PRL is known to cause hypogonadotropic hypogonadism by alteration of the gonadotropin secretory pattern (24). Moreover, PRL lev-els were not evaluated in the studies cited above. An alter-native explanation may imply the existence of a common mechanism resulting in concurrent hyperprolactinemia in CNFTs and hypogonadotropic hypogonadism, perhaps through perturbations of the hypothalamic dopaminergic pathways. The underlying mechanism(s) remain to be elucidated.
as tertiary referrals most had resistant or complicated dis-ease. Radiographic cure, defined as absence of visualized tumor, was attained in only 21–34% of tumors analyzed. The low rate of apparent tumor eradication (21%) observed for prolactinoma was predictable because the majority of these tumors were treated pharmacologically. Dopamine agonist therapy for prolactinoma generally decreases serum PRL levels, restores the eugonadal state, and decreases tumor mass. Meta-analysis of 271 macroprolactinomas revealed that 79% of patients showed shrinkage of at least 25% of tumor mass (25). Some microprolactinomas and a few larger tumors shrink to the point of disappearance, however, the majority of medically-treated prolactinomas do not. More-over, dopamine-agonist therapy is not curative. Discontin-uation of the drug usually results in recurrent hyperpro-lactinemia and often tumor reexpansion (8). In addition, the patient population included in the registry is primarily ter-tiary referral based. Indeed, 56% (n 5 249) of registered patients were referred by other endocrinologists. Thus, this patient population represents a group with more aggressive disease or disease refractory to standard specialist treatment approaches.
Intuitively, surgical resection seems more likely to com-pletely eradicate a benign tumor mass, with the caveat that surgical outcome is highly dependent on the skill and ex-pertise of the neurosurgeon. There is little literature available regarding radiographic cure rates for the hormone-secreting pituitary tumor types, as surgical success rates are generally based on biochemical outcome. Abnormal elevation of tu-mor-specific hormonal products is generally more sensitive than the radiographic visualization of tumor. Indeed, bio-chemical remission often did not correlate with radiographic cure of patients in the pituitary tumor registry. Surgical cure rates and frequency of radiological evidence for residual tumor posttherapeutically have also been poorly docu-mented for CNFT (18), even though serial radiographic im-aging is obligatory due to the lack of a reliable biochemical marker. These results suggest that the majority of CNFTs and GH-secreting tumors were incompletely excised, possibly due to tumor extension to anatomical areas that are less surgically accessible. Conversely, tumor size alone does not reliably predict likelihood for radiographic cure, as 83% of cured CNFTs (n512) and 77% of cured acromegaly tumors (n544) were macroadenomas.
Less than 40% of patients in the registry bearing functional tumors attained biochemical remission. The published sur-gical remission rate for acromegaly ranges from 42– 88% (summarized in Ref. 26), whereas the remission rate for med-ical therapy in conjunction with incomplete surgmed-ical resec-tion was 40 – 60% (27–30). Remission rates for dopamine ag-onist-treated prolactinomas ranged from 60 – 80% for macroadenomas (31–33) and 70 –90% for microprolactino-mas (34). Notably, remission rates encountered in this reg-istry for both tumor types are either at the lowest extreme or less than earlier reports. Several possibilities account for these observations. First, this tertiary-referred patient pop-ulation represents a group with more aggressive or refrac-tory disease. The high percentage of patients in the registry that required multimodal therapy lends support to this no-tion. Second, the criteria for remission were more stringent
than those used in most reported series, particularly for acromegaly. The majority of acromegaly outcome studies were based on single random GH levels ranging from 2.5–10 mg/L (30). Single serum GH measurements, in the absence of a dynamic suppression test, are not reliable indicators of disease status because GH release is pulsatile. GH concen-trations vary from undetectable (in standard assays) about half the daytime to 30mg/L in normal individuals (35, 36). In acromegaly, basal GH is rarely undetectable and overlaps unacceptably with the normal range (37). Circulating IGF-I levels, which reflect integrated 24-h GH concentrations, are a better indicator of GH hypersecretion. This test coupled with a glucose-suppressed GH is the current recommended strategy for diagnosing and monitoring acromegaly (10).
Prolonged exposure to excessive circulating GH levels is associated with an approximately 3-fold increased mortality rate (15, 38, 39). In contrast, the calculated SMR for the reg-istered acromegaly population did not indicate an increased death rate. Several recent series have confirmed that therapy resulting in biochemical remission of acromegaly normalizes mortality rate to that of the general population (26, 40, 41). Thus, it is possible that the normal mortality rate observed reflects the influence of partially normalized GH levels. Al-ternatively, the calculated mortality ratio may underestimate the true death rate. First, the current survival status could not be ascertained for 27% of patients, and deceased patients are likely to be lost to follow-up at a greater frequency. Addi-tionally, patients who died before presentation to a tertiary center did not have an opportunity to be included in the calculation. These results demonstrate the importance of suc-cessful biochemical control and appropriate long-term man-agement of these patients.
In conclusion, we have developed a novel computerized pituitary tumor registry program designed to collect com-prehensive demographic, therapeutic, pathologic, and out-come information on patients harboring these rare tumors and who are referred for tertiary consultation. Specifically, predictors of pituitary tumor recurrence and markers of per-sistent disease activity may be identified. This tool will be used prospectively to overcome the limitations introduced by the incomplete data ascertainment inherent to retrospec-tive analyses of rare diseases. This registry can be used to improve surveillance protocols and long-term therapeutic outcomes. Moreover, identification of patients with low like-lihood for recurrence should reduce the amount of healthcare resources unnecessarily consumed and improve the overall cost-effectiveness of long-term pituitary tumor management.
Acknowledgments
We acknowledge Drs. Martin Weiss and Hyrar Shahinian for gen-erously allowing us access to their patient records.
References
1.Molitch ME.1997 Pituitary incidentalomas. Endocrinol Metab Clin North Am. 26:725–740.
2.Annegers JF, Coulam CB, Laws ER.1982 Pituitary tumors: epidemiology. In: Givens JR, ed. Hormone-secreting pituitary tumors. Chicago: Year Book Med-ical Publishers; 393– 403.
3.Vance ML.1994 Hypopituitarism. N Engl J Med. 330:1651–1662. 4.Melmed S.1990 Acromegaly. N Engl J Med. 322:966 –977.
5.Etxabe J, Vazquez JA.1994 Morbidity and mortality in Cushing’s disease: an epidemiological approach. Clin Endocrinol. 40:479 – 484.
6.Snyder PJ.1997 Gonadotroph and other clinically nonfunctioning pituitary adenomas. Cancer Treat Res. 89:57–72.
7.Breslow NE, Day NE.1987 Statistical methods in cancer research. In: Vol. II—The design and analysis of cohort studies. Lyon: IARC Scientific Publica-tions. 82:65–71.
8.Molitch ME.1995 Prolactinoma. In: Melmed S, ed. The pituitary. Cambridge: Blackwell Science; 443– 477.
9.Klijn JGM, Lamberts SWJ, De Jong FH, Docter R, Van Dongen KJ, Birken-hager JC.1980 The importance of pituitary tumour size in patients with hyperprolactinaemia in relation to hormonal variables and extrasellar exten-sion of tumour. Clin Endrocrinol. 12:341–355.
10. Shimon I, Melmed S.1998 Management of pituitary tumors. Ann Intern Med. 129:472– 483.
11. Calle-Rodrigue RDP, Giannini C, Scheithauer BW,et al.1998 Prolactinomas in male and female patients: a comparative clinicopathologic study. Mayo Clin Proc. 73:1046 –1052.
12. Lloyd GM, Meares JD, Jacobi J.1975 Effects of oestrogen and bromocriptine onin vivosecretion and mitosis in prolactin cells. Nature. 255:497– 498. 13. Amara JF, Ban Itallie C, Dannies PS.1987 Regulation of prolactin production
and cell growth by estradiol: difference in sensitivity to estradiol occurs at level of messenger ribonucleic acid accumulation. Endocrinology. 133:397– 406. 14. Alexander L, Appleton D, Hall R, Ross WM, Wilkinson R.1980
Epidemi-ology of acromegaly in the Newcastle region. Clin Endocrinol. 12:71–79. 15. Rajasoorya C, Holdaway IM, Wrightson P, Scott DJ, Ibbertson HK.1994
Determinants of clinical outcome and survival in acromegaly. Clin Endocrinol. 41:95–102.
16. Frawley LS, Boockfor FR.1991 Mammosomatotrophs: presence and functions in normal and neoplastic pituitary tissue. Endocr Rev. 12:337–355. 17. Lloyd RV, Cano M, Chandler WF, Barkan AL, Horvath E, Kovacs K.1989
Human growth hormone and prolactin-secreting pituitary adenomas analyzed byin situhybridization. Am J Pathol. 134:605– 613.
18. Molitch ME.1998 Natural history and diagnosis of clinically non-functioning adenomas. In: Webb SM, ed. Pituitary tumours: epidemiology, pathogenesis, and management. Bristol: BioScientifica; 15–27.
19. Smith MV, Laws ER.1994 Magnetic resonance imaging measurements of pituitary stalk compression and deviation in patients with nonprolactin-se-creting intrasellar and parasellar tumors: lack of correlation with serum pro-lactin levels. Neurosurgery. 34:834 – 839.
20. Lees PD, Pickard JD.1987 Hyperprolactinemia, intrasellar pituitary tissue pressure, and the pituitary stalk compression syndrome. J Neurosurg. 67:192–196.
21. Lees PD, Fahlbusch R, Zrinzo A, Pickard JD.1994 Intrasellar pituitary tissue pressure, tumour size and endocrine status—an international comparison in 107 patients. Br J Neurosurg. 8:313–318.
22. Greenman Y, Tordjman K, Kisch E, Razon N, Ouaknine G, Stern N.1995 Relative sparing of anterior pituitary function in patients with growth hor-mone-secreting macroadenomas: comparison with nonfunctioning macroad-enomas. J Clin Endocrinol Metab. 80:1577–1583.
23. Damjanovic SS, Popovic VP, Petakov MS, Nikolic-Durovic MM, Doknic MZ, Gligorovic MS.1996 Gonadotrophin and free a-subunit secretion in patients with acromegaly and clinically non-functioning pituitary tumors: anterior pituitary function and the effect of thyrotrophin-releasing hormone. J Endocrinol Invest. 19:663– 669.
24. Sauder SE, Frager M, Case GD, Kelch RP, Marshall JC.1984 Abnormal patterns of pulsatile lutenizing hormone secretion in women with hyperpro-lactinemia and amenorrhea: responses to bromocriptine. J Clin Endocrinol Metab. 59:941–948.
25. Bevan JS, Webster J, Burke CW, Scanlon MF.1992 Dopamine agonists and pituitary tumour shrinkage. Endocr Rev. 13:220 –240.
26. Abosch A, Tyrrell JB, Lamborn KR, Hannegan LT, Applebury CB, Wilson CB.1998 Transsphenoidal microsurgery for growth hormone-secreting pitu-itary adenomas: initial outcome and long-term results. J Clin Endocrinol Metab. 83:3411–3418.
27. Newman C, Melmed S, George A, et al.1998 Octreotide as primary therapy for acromegaly. J Clin Endocrinol Metab. 83:3036 –3040.
28. Newman CB, Melmed S, Snyder PJ,et al.1995 Safety and efficacy of long term octreotide therapy of acromegaly: results of a multicenter trial in 103 patients. J Clin Endocrinol Metab. 80:2768 –2775.
29. Sassolas G, Harris AG, James-Deidier A.1990 French SMS 201-995 acromeg-aly study group: long-term effect of incremental doses of the somatostatin analog SMS 201-995 in 58 acromegalic patients. J Clin Endocrinol Metab. 71:391–397.
30. Vance ML, Harris AG.1991 Long-term treatment of 189 acromegalic patients with the somatostatin analog octreotide. Results of the international multi-center acromegaly study group. Arch Intern Med. 151:1573–1578.
31. Liuzzi A, Dallabonzana D, Oppizzi G,et al.1985 Low doses of dopamine agonists in the long-term treatment of macroprolactinomas. N Engl J Med. 313:656 – 659.
32. Colao A, Merola B, Sarnacchiaro F,et al.1995 Comparison among different dopamine-agonists of new formulation in the clinical management of macro-prolactinomas. Horm Res. 44:222–228.
33. Ferrari CI, Abs R, Bevan JS,et al.1997 Treatment of macroprolactionoma with cabergoline: a study of 85 patients. Clin Endocrinol. 46:409 – 413.
34. Molitch ME, Thorner MO, Wilson C.1997 Therapeutic controversy: man-agement of prolactinomas. J Clin Endocrinol Metab. 82:996 –1000.
35. Ho KY, Evans WS, Blizzard RM,et al.1987 Effects of sex and age on the 24-hour profile of growth hormone secretion in man: importance of endoge-nous estradiol concentrations. J Clin Endocrinol Metab. 64:51–58.
36. Chapman IM, Hartman ML, Straume M, Johnson ML, Veldhuis JD, Thorner MO.1994 Enhanced sensitivity growth hormone (GH) chemiluminescence assay reveals lower postglucose nadir GH concentrations in men than in women. J Clin Endocrinol Metab. 78:1312–1319.
37. Ho K, Weissberger AJ.1994 Characterization of 24-hour growth hormone secretion in acromegaly: implications for diagnosis and therapy. Clin Endo-crinol. 41:75– 83.
38. Bengtsson B-A, Eden S, Ernst I, Oden A, Sjogren B.1988 Epidemiology and long-term survival in acromegaly. Acta Med Scand. 223:327–335.
39. Bates AS, Van’t Hoff W, Jones JM, Clayton RN.1993 An audit of outcome of treatment in acromegaly. Q J Med. 86:293–299.
40. Orme SM, McNally RJQ, Cartwright RA, Belchetz PE.1998 Mortality and cancer incidence in acromegaly: a retrospective cohort study. J Clin Endocrinol Metab. 83:2730 –2734.
41. Swearingen B, Barker II FG, Katznelson L,et al.1998 Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegaly. J Clin Endocrinol Metab. 83:3419 –3426.