Genetic variants of miRNA sequences and non–small cell lung cancer survival
Full text
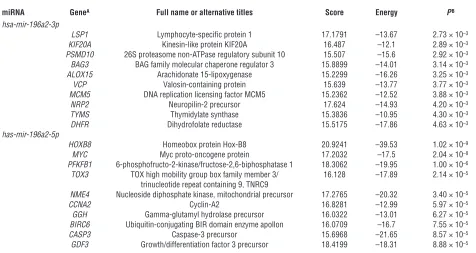
Figure

Related documents
PhotoFile IMS Mugshot/Lineup System: This software module allows the photo capture process to attach a digital color photo to inmate and master name index file with agency
We put forward two criteria to evaluate alternative policies employed by pension funds : the funding policy and allocation rules must give an ex ante fair compensation for risk taken
People living in large urban areas, especially in developing countries, where the health risks of air pollution may be underappre- ciated and pollution controls lacking, are
Average daily sunshine time, latitude, temperature and HDI After identification of the geographical location of the city or area participating in the study, we used the Hong
fectively eliminate any review of arbitration agreements under state laws of unconscionability. As a result, the Supreme Court has unwit- tingly encouraged companies to
For our HCNNs, 5 (resp. 11) multiplicative depth is required: 2 (resp. 3) ciphertext by ciphertext (in the square layers) and 3 (resp. 8) ciphertext by plaintext (in
