Acta Cryst.(2002). E58, o509±o510 DOI: 10.1107/S1600536802005792 Wolfgang Kliegelet al. C5H12NO+Clÿ
o509
organic papers
Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
N
-Hydroxypiperidinium chloride
Wolfgang Kliegel,aUlf Riebe,a
Brian O. Patrick,bSteven J.
Rettigband James Trotterb*
aInstitut fuÈr Pharmazeutische Chemie,
Technische UniversitaÈt Braunschweig, Beethovenstrasse 55, 38106 Braunschweig, Germany, andbDepartment of Chemistry, University of British Columbia, Vancouver, BC, Canada V6T 1Z1
Correspondence e-mail: brian@xray1.chem.ubc.ca
Key indicators Single-crystal X-ray study
T= 173 K
Mean(C±C) = 0.002 AÊ
Rfactor = 0.028
wRfactor = 0.061
Data-to-parameter ratio = 18.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2002 International Union of Crystallography Printed in Great Britain ± all rights reserved
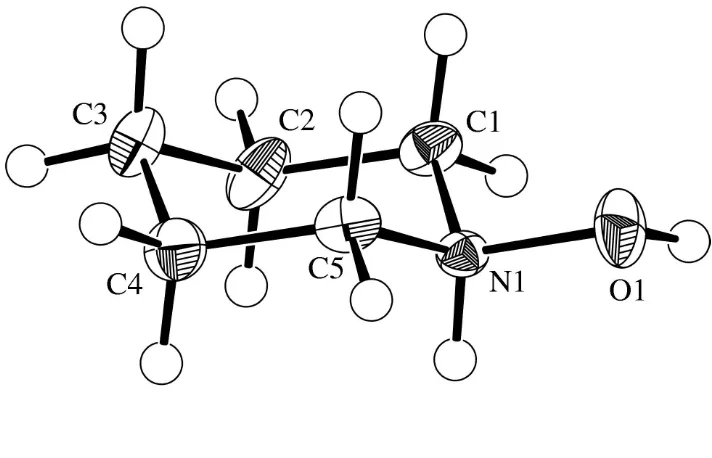
[(CH2)5NHOH]+Clÿ contains a six-membered piperidinium
ring with a chair conformation, linked to chloride ions by NÐ
H Cl and OÐH Cl hydrogen bonds. The hydroxy
substituent is in an equatorial site.
Comment
Crystals of a by-product isolated during the synthesis of a chloral adduct by reaction ofN-hydroxypiperidine and chloral hydrate (Zinneret al., 1965; Kliegelet al., 2001) proved to be
N-hydroxypiperidine hydrochloride (N-hydroxypiperidinium chloride), (I) (Fig. 1). The salt, which has been well known for a long time (Wernick & Wolffenstein, 1898; Thesing & Mayer, 1956), probably originates from the formation of HCl during the reaction by partial decomposition of chloral hydrate, the mechanism of which is not clear. The presence of water and the basic reagent (N-hydroxypiperidine) might produce HCl and dichloroacetic acid, or chloroform, which could be the source for HCl (Fairbrother, 1973; Lutnitskii, 1975).
The cation contains a six-membered piperidinium ring with a normal chair conformation (dihedral angle magnitudes 56.4± 57.8), and the hydroxy substituent in the equatorial site. Bond
lengths and angles differ slightly from those in piperidinium chloride (ReÂrat, 1960; Dattagupta & Saha, 1975; Gaudetet al., 1989). In particular, there is some asymmetry in the molecular dimensions as a result of the presence of the OH H atom, which has a staggered conformation about the NÐO bond. The two OÐNÐC angles differ signi®cantly [111.4 (1) and 106.3 (1)], the distortion presumably resulting from
intra-molecular (the OH H atom is on the side of the larger angle) or intermolecular (hydrogen bonds) steric interactions. There is also a slight alternation in the values of the endocyclic bond angles, with those at C1, C3, and C5 being exactly tetrahedral [109.6 (1)] and those at C2, C4 [111.3 (1)] and especially at N
[112.9 (1)] being slightly larger. The NÐO bond length
[1.418 (2) AÊ] is similar to that in protonated hydroxylamine (H2NOHHCl) [1.411 (2) AÊ; Shiet al., 1987].
510
Two cations and twoanions are linked about a centre of inversion by OÐH Cl and NÐH Cl hydrogen bonds, to produce a ten-membered hydrogen-bonded ring: O Cl = 2.967 (1), OÐH = 0.92 (2), H Cl = 2.05 (2) AÊ, OÐH Cl = 170 (2); N Cl = 3.044 (1),
NÐH = 0.91 (2), H Cl = 2.14 (2) AÊ, NÐH Cl = 170 (1);
O Cl N = 112.0 (1). These units are linked by weaker
(van der Waals) forces, with a possible intermolecular CÐ H O bond, C O = 3.374 (2), H O = 2.44 AÊ, CÐH O = 159, and a possible CÐH Cl bond [C Cl = 3.624 (2),
H Cl = 2.71 AÊ and CÐH Cl = 156].
Experimental
Crystals were obtained as a by-product of the reaction of chloral hydrate andN-hydroxypiperidine (Kliegelet al., 2001).
Crystal data C5H12NO+Clÿ Mr= 137.61 Monoclinic,P21=c a= 7.1304 (5) AÊ b= 7.0213 (5) AÊ c= 14.4857 (9) AÊ
= 93.333 (4) V= 724.00 (7) AÊ3 Z= 4
Dx= 1.262 Mg mÿ3
Mo Kradiation Cell parameters from 4174
re¯ections
= 2.9±27.8
= 0.44 mmÿ1 T= 173 K Block, colorless 0.200.200.20 mm Data collection
Quantum CCD diffractometer
'and!scans
Absorption correction: multi-scan (d*TREK; Molecular Structure Corporation, 2001)
Tmin= 0.86,Tmax= 0.92
6650 measured re¯ections
1526 independent re¯ections 1230 re¯ections withI> 3(I) Rint= 0.029
max= 27 h=ÿ8!9 k=ÿ8!8 l=ÿ17!15 Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.028 wR(F2) = 0.061 S= 1.63 1526 re¯ections 81 parameters
H atoms treated by a mixture of independent and constrained re®nement
w= 1/[2(Fo)]
(/)max= 0.001
max= 0.26 e AÊÿ3
min=ÿ0.29 e AÊÿ3
Table 1
Selected geometric parameters (AÊ,).
O1ÐN1 1.418 (2)
N1ÐC1 1.484 (2)
N1ÐC5 1.490 (2)
C1ÐC2 1.515 (2)
C2ÐC3 1.524 (2)
C3ÐC4 1.516 (2)
C4ÐC5 1.514 (2)
O1ÐN1ÐC1 111.4 (1)
O1ÐN1ÐC5 106.3 (1)
C1ÐN1ÐC5 112.9 (1)
N1ÐC1ÐC2 109.6 (1)
C1ÐC2ÐC3 111.2 (1)
C2ÐC3ÐC4 109.6 (1)
C3ÐC4ÐC5 111.3 (1)
N1ÐC5ÐC4 109.6 (1)
Data collection: d*TREK (Molecular Structure Corporation, 2001); cell re®nement: d*TREK; data reduction: d*TREK; program(s) used to solve structure: SIR97 (Altomare et al., 1999); program(s) used to re®ne structure:TEXSAN(Molecular Structure Corporation, 1997); software used to prepare material for publica-tion:TEXSAN.
We thank the Natural Sciences and Engineering Research Council of Canada and the Fonds der Chemische Industrie, Frankfurt am Main, for ®nancial support
References
Altomare, A., Burla, M. C., Camalli, M., Cascarano, G., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999).J. Appl. Cryst.32, 115±119.
Dattagupta, J. K. & Saha, N. N. (1975).J. Cryst. Mol. Struct.5, 177±189. Fairbrother, J. E. (1973).Analytical Pro®les of Drug Substances, Vol. 2, edited
by K. Florey, pp. 85±143. New York: Academic Press.
Gaudet, M. V., Zaworotko, M. J. & White, P. S. (1989).Inorg. Chem.28, 1191± 1193.
Kliegel, W., Riebe, U., Patrick, B. O., Rettig, S. J. & Trotter, J. (2001).Acta Cryst.E57, o1173±o1174.
Lutnitskii, F. I. (1975).Chem. Rev.75, 259±290.
Molecular Structure Corporation (1997).TEXSAN.Version 1.8. MSC, 3200 Research Forest Drive, The Woodlands, TX 77381, USA.
Molecular Structure Corporation (2001).d*TREK. Version 7.11. MSC, 3200 Research Forest Drive, The Woodlands, TX 77381, USA.
ReÂrat, C. (1960).Acta Cryst.13, 72±80.
Shi, K.-L., Wang, R.-Q. & Mak, T. C. W. (1987).J. Mol. Struct.160, 109±116. Thesing, I. & Mayer, H. (1956).Chem. Ber.89, 2159±2167.
Wernick, W. & Wolffenstein, R. (1898).Ber. Dtsch. Chem. Ges.31, 1553±1561. Zinner, G., Ritter, W. & Kliegel, W. (1965).Pharmazie,20, 291±296. Figure 1
supporting information
sup-1
Acta Cryst. (2002). E58, o509–o510supporting information
Acta Cryst. (2002). E58, o509–o510 [doi:10.1107/S1600536802005792]
N
-Hydroxypiperidinium chloride
Wolfgang Kliegel, Ulf Riebe, Brian O. Patrick, Steven J. Rettig and James Trotter
S1. Comment
Crystals of a by-product isolated during the synthesis of a chloral adduct by reaction of N-hydroxypiperidine and chloral
hydrate (Zinner et al., 1965; Kliegel et al., 2001) proved to be N-hydroxypiperidine hydrochloride (N
-hydroxy-piperidinium chloride), (I) (Fig. 1). The salt, which has been well known for a long time (Wernick & Wolffenstein, 1898;
Thesing & Mayer, 1956), probably originates from the formation of HCl during the reaction by partial decomposition of
chloral hydrate, the mechanism of which is not clear. The presence of water and the basic reagent (N-hydroxypiperidine)
might produce HCl and dichloroacetic acid, or chloroform which could be the source for HCl (Fairbrother, 1973;
Lutnitskii, 1975).
The cation contains a six-membered piperidinium ring with a normal chair conformation (dihedral angle magnitudes
56.4–57.8°), and the hydroxy substituent in the equatorial site. Bond lengths and angles differ slightly from those in
piperidinium chloride (Rérat, 1960; Dattagupta & Saha, 1975; Gaudet et al., 1989). In particular, there is some
asymmetry in the molecular dimensions as a result of the presence of the OH H atom, which has a staggered
conformation about the N—O bond. The two O—N—C angles differ significantly [111.4 (1) and 106.3 (1)°], the
distortion presumably resulting from intramolecular (the OH H atom is on the side of the larger angle) or intermolecular
(hydrogen bonds) steric interactions. There is also a slight alternation in the values of the endocyclic bond angles, with
those at C1, C3, and C5 being exactly tetrahedral [109.6 (1)°], and those at C2, C4 [111.3 (1)°] and especially at N
[112.9 (1)°] being slightly larger. The N—O bond length, 1.418 (2) Å, is similar to that in protonated hydroxylamine
(H2NOH·HCl), 1.411 (2) Å (Shi et al., 1987).
Two cations and two anions are linked about a centre of inversion by O—H···Cl and N—H···Cl hydrogen bonds, to
produce a ten-membered hydrogen-bonded ring: O···Cl = 2.967 (1), O—H = 0.92 (2), H···Cl = 2.05 (2) Å, O—H···Cl =
170 (2)°; N···Cl = 3.044 (1), N—H = 0.91 (2), H···Cl = 2.14 (2) Å, N—H···Cl = 170 (1)°; O···Cl···N = 112.0 (1)°. These
units are linked by weaker (van der Waals) forces, with a possible intermolecular C—H···O bond, C···O = 3.374 (2), H···O
= 2.44 Å, C—H···O = 159°, and a possible C—H···Cl bond [C···Cl = 3.624 (2), H···Cl = 2.71 Å and C—H···Cl = 156°].
S2. Experimental
Figure 1
View of the title structure shown with 50% ellipsoids.
(I)
Crystal data C5H12NO+·Cl−
Mr = 137.61 Monoclinic, P21/c
a = 7.1304 (5) Å b = 7.0213 (5) Å c = 14.4857 (9) Å β = 93.333 (4)° V = 724.00 (7) Å3
Z = 4
F(000) = 296 Dx = 1.262 Mg m−3
Mo Kα radiation, λ = 0.7107 Å Cell parameters from 4174 reflections θ = 2.9–27.8°
µ = 0.44 mm−1
T = 173 K Block, colorless 0.20 × 0.20 × 0.20 mm
Data collection Quantum CCD diffractometer
Radiation source: X-ray tube Graphite monochromator area detector scans
Absorption correction: multi-scan
(D*TREK; Molecular Structure Corporation,
2001)
Tmin = 0.86, Tmax = 0.92
6650 measured reflections 1526 independent reflections 1230 reflections with I > 3σ(I) Rint = 0.029
θmax = 27°, θmin = 2.9°
h = −8→9
k = −8→8
supporting information
sup-3
Acta Cryst. (2002). E58, o509–o510Refinement Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.028
wR(F2) = 0.061
S = 1.63 1526 reflections 81 parameters 0 restraints
0 constraints
H atoms treated by a mixture of independent and constrained refinement
Weighting scheme based on measured s.u.'s w = 1/[σ2(F
o)]
(Δ/σ)max = 0.001
Δρmax = 0.26 e Å−3
Δρmin = −0.29 e Å−3
Special details
Experimental. Data were collected in 0.50° oscillations with 60.0 s exposures. A sweep of data was done using φ oscillations from 0.0 to 190.0° at χ = -90° and a second sweep was performed using ω oscillations between -23.0 and 18.0° at χ = -90.0°. The crystal-to-detector distance was 38.83 mm. The detector swing angle was -10.0°. The absorption correction is based on a three-dimensional analysis of symmetry-equivalent data and is performed along with batch scaling in a single step.
Refinement. H11 and H12 (bonded to N and O respectively) were refined isotropically; the other H atoms were fixed in idealized sites.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Cl1 0.24319 (5) 0.09979 (6) 1.08369 (2) 0.0288 (1)
O1 0.1190 (2) −0.0039 (2) 0.83651 (7) 0.0312 (3)
N1 0.1981 (2) 0.1649 (2) 0.87582 (8) 0.0177 (3)
C1 0.0638 (2) 0.3262 (2) 0.86723 (9) 0.0237 (4)
C2 0.1571 (2) 0.5054 (2) 0.9058 (1) 0.0289 (4)
C3 0.3365 (2) 0.5492 (2) 0.8576 (1) 0.0298 (4)
C4 0.4680 (2) 0.3798 (2) 0.8663 (1) 0.0285 (4)
C5 0.3741 (2) 0.2008 (2) 0.82795 (9) 0.0231 (4)
H1 0.0249 0.3458 0.8019 0.028*
H2 −0.0468 0.2972 0.9019 0.028*
H3 0.0698 0.6125 0.8967 0.035*
H4 0.1880 0.4879 0.9720 0.035*
H5 0.3053 0.5756 0.7921 0.036*
H6 0.3980 0.6609 0.8865 0.036*
H7 0.5796 0.4062 0.8321 0.034*
H8 0.5055 0.3597 0.9318 0.034*
H9 0.4595 0.0924 0.8379 0.028*
H10 0.3438 0.2167 0.7615 0.028*
H11 0.227 (2) 0.142 (3) 0.937 (1) 0.044 (5)*
H12 0.007 (3) −0.020 (3) 0.865 (1) 0.067 (7)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Cl1 0.0277 (2) 0.0386 (2) 0.0195 (2) −0.0037 (2) −0.0034 (1) 0.0078 (2)
O1 0.0382 (7) 0.0242 (6) 0.0317 (6) −0.0101 (6) 0.0064 (5) −0.0127 (5)
N1 0.0217 (6) 0.0176 (6) 0.0137 (6) 0.0008 (5) 0.0010 (4) −0.0006 (4)
C2 0.0330 (9) 0.0173 (8) 0.0379 (9) 0.0047 (8) 0.0143 (7) 0.0035 (7)
C3 0.0321 (9) 0.0237 (9) 0.0344 (9) −0.0055 (7) 0.0078 (7) 0.0043 (6)
C4 0.0203 (8) 0.035 (1) 0.0306 (8) −0.0022 (8) 0.0042 (6) 0.0058 (7)
C5 0.0197 (7) 0.0282 (9) 0.0221 (7) 0.0066 (7) 0.0064 (5) 0.0018 (6)
Geometric parameters (Å, º)
O1—N1 1.418 (2) C2—H4 0.98
O1—H12 0.92 (2) C3—C4 1.516 (2)
N1—C1 1.484 (2) C3—H5 0.98
N1—C5 1.490 (2) C3—H6 0.98
N1—H11 0.91 (2) C4—C5 1.514 (2)
C1—C2 1.515 (2) C4—H7 0.98
C1—H1 0.98 C4—H8 0.98
C1—H2 0.98 C5—H9 0.98
C2—C3 1.524 (2) C5—H10 0.98
C2—H3 0.98
CL1···O1i 2.967 (1) O1···C3iii 3.506 (2)
CL1···N1 3.044 (1) O1···C2iii 3.595 (2)
O1···C1ii 3.374 (2)
N1—O1—H12 105 (1) C2—C3—C4 109.6 (1)
O1—N1—C1 111.4 (1) C2—C3—H5 109.4
O1—N1—C5 106.3 (1) C2—C3—H6 109.4
O1—N1—H11 108 (1) C4—C3—H5 109.4
C1—N1—C5 112.9 (1) C4—C3—H6 109.4
C1—N1—H11 109 (1) H5—C3—H6 109.5
C5—N1—H11 109 (1) C3—C4—C5 111.3 (1)
N1—C1—C2 109.6 (1) C3—C4—H7 109.0
N1—C1—H1 109.4 C3—C4—H8 109.0
N1—C1—H2 109.4 C5—C4—H7 109.0
C2—C1—H1 109.4 C5—C4—H8 109.0
C2—C1—H2 109.4 H7—C4—H8 109.5
H1—C1—H2 109.5 N1—C5—C4 109.6 (1)
C1—C2—C3 111.2 (1) N1—C5—H9 109.4
C1—C2—H3 109.0 N1—C5—H10 109.4
C1—C2—H4 109.0 C4—C5—H9 109.4
C3—C2—H3 109.0 C4—C5—H10 109.4
C3—C2—H4 109.0 H9—C5—H10 109.5
H3—C2—H4 109.5
O1—N1—C1—C2 −177.3 (1) C1—N1—C5—C4 57.7 (1)
O1—N1—C5—C4 −179.9 (1) C1—C2—C3—C4 −56.4 (2)
N1—C1—C2—C3 56.6 (2) C2—C1—N1—C5 −57.8 (1)
N1—C5—C4—C3 −56.5 (2) C2—C3—C4—C5 56.4 (2)