metal-organic papers
m580
Md. Khayrul Kabiret al. [Ni(C2H5N4S)2] DOI: 10.1107/S1600536802017038 Acta Cryst.(2002). E58, m580±m582 Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
cis
-Bis(amidinothioureato-
j
2N,N
000)nickel(II)
Md. Khayrul Kabir,aKoichi Yamada,bKeiichi Adachi,b Mitsuru Kondoaand Satoshi Kawatab*
aDepartment of Chemistry, Shizuoka University,
Ohya, Shizuoka 422-8529, Japan, and
bDepartment of Chemistry, Graduate School of
Science, Osaka University, Machikaneyamacho, Toyonaka, Osaka 560-0043, Japan
Correspondence e-mail: kawata@chem.sci.osaka-u.ac.jp
Key indicators Single-crystal X-ray study
T= 295 K
Mean(N±C) = 0.006 AÊ
Rfactor = 0.053
wRfactor = 0.134
Data-to-parameter ratio = 13.6
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2002 International Union of Crystallography Printed in Great Britain ± all rights reserved
The title compound, [Ni(C2H5N4S)2], has a twofold axis. The NiII atom is coordinated in a deformed square-planar geometry by four imino N atoms of two atu ligands (Hatu = amidinothiourea). Two six-membered chelate rings including the Ni atom are twisted with a dihedral angle of 17.24 (16).
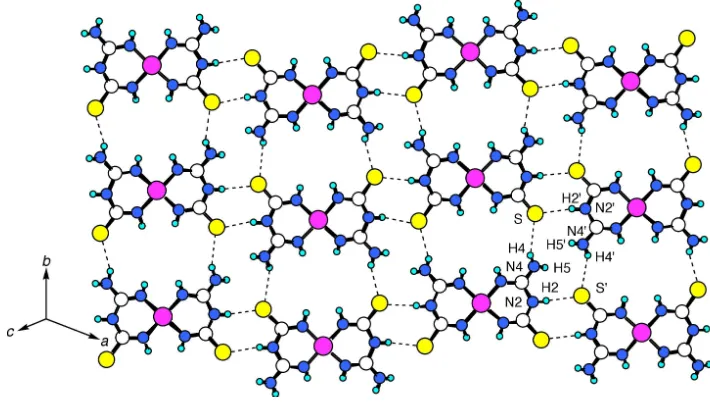
The [Ni(atu)2] units make a two-dimensional layer via NÐ H S hydrogen bonds.
Comment
Amidinothiourea (Hatu) can be used to construct a metal-complex-based module for superstructures as it has available coordination sites (Vilar et al., 1998). Moreover, SN donors stabilize the lower oxidation states of metal atoms and lower the electron density at NO in metal nitrosyls (Chakrabartyet al., 1990).
Hatu has two tautomeric forms (see Scheme). It can coordinate to metal ions using either two N atoms (N,N0
-chelating) or one N and one S atom (N,S-chelating). However, very few reports have been appeared on the Hatu ligand (Vilaret al., 1998, 1999; Chenget al., 2001). Among them one report shows that Hatu is coordinated to NiIIion in anN,N0
-chelating mode, to form atranscomplex (Vilaret al., 1999). During our studies on coordination compounds with Hatu, orange single crystals of the title compound, (I), were obtained. The crystal structure of (I) is presented here.
The crystal structure of (I) consists of a mononuclear complex Ni(atu)2. AnORTEP-3 (Farrugia, 1997) drawing with the atom-numbering scheme is shown in Fig. 1. The NiIIatom is coordinated in a deformed square-planar geometry by four imino N atoms of two atu ligands. The two six-membered rings incorporating the Ni atom are twisted with a dihedral angle of 17.24 (16). The NiÐN distances are 1.860 (4) and 1.855 (4) AÊ
(Table 1). The shortness of the CÐN bonds (1.29±1.37 AÊ) indicates the presence of multiple bonding with delocalization ofelectrons. The coordination geometry of (I) is similar to that of [Pd(atu)2]Cl21.5H2O (Chakrabartyet al., 1990), but the coordinating atoms (N,S-chelating) are different in that case. On the other hand,trans-Ni(atu)2has the same coordinating atoms (N,N0-chelating) with similar NiÐN distances (Vilaret
al., 1999), but the crystal packing modes of the two compounds are different. Theciscoordination mode in (I) has multi-site hydrogen-bonding ability that allows the mononuclear building module to form a hydrogen-bond-supported two-dimensional layer structure. Ni(atu)2molecules are connected to each other via hydrogen-bonding interactions, which are formed between terminal amino groups and S atoms, gener-ating a straight tape along thebaxis. The tapes are hydrogen bonded to form a two-dimensional layer perpendicular to the [101] direction (Fig. 2 and Table 2). The layers make a three-dimensional packing structure through stacking interactions (Fig. 3). The minimum distance between the layers (C N) is 3.319 (6) AÊ.
Experimental
To a solution of nickel acetate monohydrate (0.019 g, 0.1 mmol) in H2O (5 ml), amidininothiourea (0.0236 g, 0.2 mmol) in methanol
(5 ml) was added without mixing the two solutions. Orange crystals of (I) began to form at ambient temperature in two weeks. One of these crystals was used for X-ray crystallography. Calculated for C4H10N8NiS2: C 16.39, H 3.44, N 38.24%; found: C 16.35, H 3.36, N
37.93%. Crystal data
[Ni(C2H5N4S)2] Mr= 293.03 Monoclinic,C2=c a= 14.804 (2) AÊ b= 7.8988 (14) AÊ c= 9.1216 (13) AÊ
= 111.732 (12) V= 990.8 (3) AÊ3 Z= 4
Dx= 1.964 Mg mÿ3 MoKradiation Cell parameters from 20
re¯ections
= 2.5±10
= 2.36 mmÿ1 T= 295 (2) K Block, orange 0.20.10.1 mm
Data collection
MacScience MXC3 diffractometer
!scans
Absorption correction: scan (Northet al., 1968) Tmin= 0.753,Tmax= 0.790 1257 measured re¯ections 1141 independent re¯ections 906 re¯ections withI> 2(I)
Rint= 0.013
max= 27.5 h=ÿ19!17 k= 0!10 l= 0!11
3 standard re¯ections every 100 re¯ections intensity decay: none
Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.053 wR(F2) = 0.134 S= 0.99 1140 re¯ections 84 parameters
Only coordinates of H atoms re®ned
w= 1/[2(F
o2) + (0.0696P)2] whereP= (Fo2+ 2Fc2)/3 (/)max= 0.001
max= 0.80 e AÊÿ3 min=ÿ0.66 e AÊÿ3
Table 1
Selected geometric parameters (AÊ,).
Ni1ÐN3 1.855 (4)
Ni1ÐN1 1.860 (4)
S1ÐC2 1.726 (4)
N1ÐC1 1.288 (6)
N2ÐC2 1.360 (6)
N2ÐC1 1.368 (5)
N3ÐC2 1.302 (6)
N4ÐC1 1.350 (6)
N3ÐNi1ÐN1 90.52 (17)
N1ÐNi1ÐN1i 90.6 (2)
N3ÐNi1ÐN3i 89.9 (2)
C1ÐN1ÐNi1 129.2 (3)
C2ÐN2ÐC1 126.5 (4)
C2ÐN3ÐNi1 130.6 (3)
N1ÐC1ÐN4 124.6 (4)
N1ÐC1ÐN2 121.7 (4)
N4ÐC1ÐN2 113.7 (4)
N3ÐC2ÐN2 119.6 (4)
N3ÐC2ÐS1 124.0 (4)
N2ÐC2ÐS1 116.4 (3)
Symmetry code: (i) 2ÿx;y;3 2ÿz.
Acta Cryst.(2002). E58, m580±m582 Md. Khayrul Kabiret al. [Ni(C2H5N4S)2]
m581
metal-organic papers
Figure 2
The layer structure of (I). H atoms have been omitted for clarity. The dashed lines represent hydrogen bonds.
Figure 1
ORTEP-3 (Farrugia, 1997) drawing of (I) with the atom-numbering scheme. Displacement ellipsoids are drawn at the 50% probability level.
Figure 3
metal-organic papers
m582
Md. Khayrul Kabiret al. [Ni(C2H5N4S)2] Acta Cryst.(2002). E58, m580±m582Table 2
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
N4ÐH4 S1i 0.77 (6) 2.77 (6) 3.434 (5) 145 (5)
N2ÐH2 S1ii 0.87 (6) 2.51 (6) 3.362 (4) 167 (5)
Symmetry codes: (i)x;yÿ1;z; (ii)5
2ÿx;12ÿy;1ÿz.
The H-atom coordinates were re®ned with a ®xedUiso value of
0.031 AÊ2.
Data collection: CRYSTAN G (MacScience, 1992); cell re®ne-ment: CRYSTAN G; data reduction: CrystalStructure (Molecular Structure Corporation & Rigaku, 2002); program(s) used to solve structure:SHELXS97 (Sheldrick, 1997); program(s) used to re®ne structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEP-3 for Windows(Farrugia, 1997); software used to prepare material for publication:SHELXL97.
This research was supported by Grants-in-Aid for Scienti®c Research on Priority Areas (No. 12023216, Metal-assembled Complexes, and No. 13031038, Dynamic Control of Strongly Correlated Soft Materials) from the Ministry of Education, Science, Sports and Culture, Japan.
References
Chakrabarty, K., Kar, T. & Gupta, S. P. S. (1990).Acta Cryst.C46, 2065±2068. Cheng, S.-T., Doxiadi, E., Vilar, R., White, A. J. P. & Williams, D. J. (2001).J.
Chem. Soc. Dalton Trans.pp. 2239±2244. Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
MacScience (1992).CRYSTAN G. MacScience Co. Ltd, Yokohama, Japan. Molecular Structure Corporation & Rigaku (2002).CrystalStructure.
Mole-cular Structure Corporation, 9009 New Trails Drive, The Woodlands, TX 77381-5209, USA, and Rigaku Corporation, Akishima, Tokyo, Japan. North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst.A24, 351±
359.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of GoÈttingen, Germany.
Vilar, R., Mingos, D. M. P., White, A. J. P. & Williams, D. J. (1998).Angew Chem. Int. Ed.37, 1258±1261.
supporting information
sup-1
Acta Cryst. (2002). E58, m580–m582
supporting information
Acta Cryst. (2002). E58, m580–m582 [doi:10.1107/S1600536802017038]
cis
-Bis(amidinothioureato-
κ
2N,N
′
)nickel(II)
Md. Khayrul Kabir, Koichi Yamada, Keiichi Adachi, Mitsuru Kondo and Satoshi Kawata
S1. Comment
Amidinothiourea (Hatu) can be used to construct a metal complex based module for supra-structures as it has available
coordination sites (Vilar et al., 1998). Moreover, SN donors stabilize the lower oxidation states of metal atoms and lower
the electron density at NO in metal nitrosyls (Chakrabarty et al., 1990). Hatu has two tautomeric forms (see Scheme). It
can coordinate to metal ions using either two N atoms (N,N′-chelating) or one N and one S atom (N,S-chelating).
However, very few reports have been appeared on the Hatu ligand (Vilar et al., 1998, 1999; Cheng et al., 2001). Among
them one report shows that Hatu is coordinated to NiII ion by N,N′-chelating mode in the trans form (Vilar et al., 1999).
During our studies on coordination compounds with Hatu, orange single crystals of the title compound, (I), were
obtained. The crystal structure of (I) is presented here.
The crystal structure of (I) consists of a mononuclear complex Ni(atu)2. An ORTEP-3 (Farrugia, 1997) drawing with the
atom-numbering scheme is shown in Fig. 1. The NiII atom is coordinated with a deformed square-planar geometry, by
four imino N atoms of two atu ligands. The two six-membered rings incorporating the Ni atom are twisted with a dihedral
angle of 17.24 (16)°. The Ni—N distances are 1.860 (4) and 1.855 (4) Å (Table 1). The shortness of the C—N bonds
(1.29–1.37 Å) indicates the presence of multiple bonding with delocalization of π electrons. This may be due to the
interactions of H atoms on the coordinated N atoms. The coordination geometry of (I) is similar to that of
[Pd(atu)2]Cl2·1.5H2O (Chakrabarty et al., 1990), but the coordinating atoms (N,S-chelating) are different. On the other
hand, trans-Ni(atu)2 has the same coordinating atoms (N,N′-chelating) with similar Ni—N distances (Vilar et al., 1999),
but the crystal packing modes of the two compounds are different. The cis coordination mode in (I) has multi-site
hydrogen-bonding ability that allows the mononuclear building module to form a hydrogen-bond-supported
two-dimensional layer structure. Ni(atu)2 molecules are connected to each other via hydrogen-bonding interactions, which are
formed between terminal amino groups and S atoms, forming a straight tape along the b direction. The tapes are
hydrogen bonded to form a two-dimensional layer perpendicular to the [101] direction (Fig. 2 and Table 2). The layers
make a three-dimensional packing structure through stacking interactions (Fig. 3). The minimum distance between the
layers (C—N) is 3.319 (6) Å.
S2. Experimental
To a solution of nickel acetate monohydrate (0.019 g, 0.1 mmol) in H2O (5 ml), amidininothiourea (0.0236 g, 0.2 mmol)
in methanol (5 ml) was added without mixing the two solutions. Orange crystals of (I) began to form at ambient
temperature in two weeks. One of these crystals was used for X-ray crystallography. Calculated for C4H10N8NiS2: C
supporting information
sup-2
Acta Cryst. (2002). E58, m580–m582
S3. Refinement
[image:5.610.120.485.102.355.2]The H-atom coordinates were refined with a fixed Ueq value of 0.031 Å2.
Figure 1
ORTEP-3 (Farrugia, 1997) drawing of (I) with the atom-numbering scheme. Displacement ellipsoids are drawn at the
50% probability level.
Figure 2
[image:5.610.125.480.408.609.2]supporting information
sup-3
[image:6.610.127.484.71.273.2]Acta Cryst. (2002). E58, m580–m582
Figure 3
Projection of the crystal structure of (I) along the b axis.
cis-bis(amidinothioureato-N,N)2Nickel(II)
Crystal data
[Ni(C2H5N4S)2] Mr = 293.03
Monoclinic, C2/c Hall symbol: -C 2yc a = 14.804 (2) Å b = 7.8988 (14) Å c = 9.1216 (13) Å β = 111.732 (12)° V = 990.8 (3) Å3 Z = 4
F(000) = 600 Dx = 1.964 Mg m−3
Mo Kα radiation, λ = 0.71069 Å Cell parameters from 20 reflections θ = 2.5–10°
µ = 2.36 mm−1 T = 295 K Block, orange 0.2 × 0.1 × 0.1 mm
Data collection
MXC3
diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω scans
Absorption correction: ψ scan (North et al., 1968)
Tmin = 0.753, Tmax = 0.790 1257 measured reflections
1141 independent reflections 906 reflections with I > 2σ(I) Rint = 0.013
θmax = 27.5°, θmin = 3.0° h = −19→17
k = 0→10 l = 0→11
3 standard reflections every 100 reflections intensity decay: none
Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.053 wR(F2) = 0.134 S = 0.99 1140 reflections 84 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
supporting information
sup-4
Acta Cryst. (2002). E58, m580–m582
w = 1/[σ2(Fo2) + (0.0696P)2] where P = (Fo2 + 2Fc2)/3 (Δ/σ)max = 0.001
Δρmax = 0.80 e Å−3 Δρmin = −0.66 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Full-matrix least-squares refinement was carried out with anisotropic thermal parameters for all non-hydrogen atoms. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Ni1 1.0000 0.10583 (9) 0.7500 0.0192 (3)
S1 1.13295 (9) 0.42264 (15) 0.47612 (16) 0.0320 (4)
N1 1.0695 (3) −0.0598 (5) 0.6914 (4) 0.0229 (8)
N2 1.1584 (3) 0.1121 (5) 0.5882 (5) 0.0226 (8)
N3 1.0518 (3) 0.2721 (5) 0.6605 (5) 0.0232 (8)
N4 1.1902 (3) −0.1706 (6) 0.6115 (6) 0.0314 (10)
C1 1.1363 (3) −0.0425 (6) 0.6339 (5) 0.0209 (9)
C2 1.1110 (3) 0.2603 (5) 0.5852 (5) 0.0218 (9)
H1 1.064 (4) −0.152 (7) 0.714 (6) 0.031*
H2 1.207 (4) 0.111 (6) 0.556 (6) 0.031*
H3 1.029 (4) 0.375 (6) 0.657 (6) 0.031*
H4 1.174 (4) −0.262 (8) 0.619 (6) 0.031*
H5 1.241 (4) −0.165 (7) 0.607 (6) 0.031*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Ni1 0.0237 (4) 0.0154 (4) 0.0226 (4) 0.000 0.0134 (3) 0.000
S1 0.0369 (7) 0.0221 (6) 0.0484 (8) 0.0055 (5) 0.0291 (6) 0.0113 (5)
N1 0.029 (2) 0.0164 (18) 0.026 (2) 0.0017 (16) 0.0137 (16) 0.0038 (15)
N2 0.0240 (19) 0.0200 (19) 0.0293 (19) 0.0018 (15) 0.0165 (16) 0.0039 (16)
N3 0.030 (2) 0.0154 (17) 0.030 (2) 0.0024 (15) 0.0185 (16) −0.0005 (16)
N4 0.034 (2) 0.019 (2) 0.049 (3) 0.0051 (18) 0.024 (2) 0.0000 (19)
C1 0.024 (2) 0.018 (2) 0.021 (2) 0.0021 (17) 0.0076 (18) −0.0010 (17)
C2 0.024 (2) 0.018 (2) 0.024 (2) −0.0029 (17) 0.0098 (18) −0.0013 (17)
Geometric parameters (Å, º)
Ni1—N3 1.855 (4) N2—H2 0.87 (6)
Ni1—N1 1.860 (4) N3—C2 1.302 (6)
S1—C2 1.726 (4) N3—H3 0.87 (5)
supporting information
sup-5
Acta Cryst. (2002). E58, m580–m582
N1—H1 0.76 (5) N4—H4 0.77 (6)
N2—C2 1.360 (6) N4—H5 0.77 (6)
N2—C1 1.368 (5)
N3—Ni1—N1 90.52 (17) Ni1—N3—H3 118 (4)
N1—Ni1—N1i 90.6 (2) C1—N4—H4 118 (4)
N3—Ni1—N3i 89.9 (2) C1—N4—H5 128 (4)
C1—N1—Ni1 129.2 (3) H4—N4—H5 113 (6)
C1—N1—H1 112 (4) N1—C1—N4 124.6 (4)
Ni1—N1—H1 118 (4) N1—C1—N2 121.7 (4)
C2—N2—C1 126.5 (4) N4—C1—N2 113.7 (4)
C2—N2—H2 119 (3) N3—C2—N2 119.6 (4)
C1—N2—H2 114 (3) N3—C2—S1 124.0 (4)
C2—N3—Ni1 130.6 (3) N2—C2—S1 116.4 (3)
C2—N3—H3 112 (4)
N3—Ni1—N1—C1 8.9 (4) C2—N2—C1—N1 −5.2 (7)
N1i—Ni1—N1—C1 179.5 (5) C2—N2—C1—N4 174.4 (4)
N3i—Ni1—N3—C2 172.2 (5) Ni1—N3—C2—N2 −12.4 (7)
N1—Ni1—N3—C2 1.7 (4) Ni1—N3—C2—S1 166.6 (2)
Ni1—N1—C1—N4 172.2 (4) C1—N2—C2—N3 15.3 (7)
Ni1—N1—C1—N2 −8.3 (7) C1—N2—C2—S1 −163.7 (4)
Symmetry code: (i) −x+2, y, −z+3/2.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N4—H4···S1ii 0.77 (6) 2.77 (6) 3.434 (5) 145 (5)
N2—H2···S1iii 0.87 (6) 2.51 (6) 3.362 (4) 167 (5)