Acta Cryst.(2002). E58, o1083±o1084 DOI: 10.1107/S1600536802015969 Hiroyuki Ishidaet al. C7H4ClNO4C6H5N3
o1083
organic papers
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
The 1:1 complex of 2-chloro-4-nitrobenzoic
acid and 1,2,3-benzotriazole
Hiroyuki Ishida,* Takeo Fukunaga and Setsuo Kashino
Department of Chemistry, Faculty of Science, Okayama University, Okayama 700-8530, Japan
Correspondence e-mail: [email protected]
Key indicators Single-crystal X-ray study T= 300 K
Mean(C±C) = 0.004 AÊ Rfactor = 0.052 wRfactor = 0.132
Data-to-parameter ratio = 13.3
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2002 International Union of Crystallography Printed in Great Britain ± all rights reserved
In the title compound, C7H4ClNO4C6H5N3, two acid and two
base components are connected by OÐH N and NÐH O hydrogen bonds to afford a centrosymmetric macrocycle with graph-set descriptor R44(16). CÐH O hydrogen bonds
connect the ring units to form a ribbon structure.
Comment
The title compound, (I), was investigated as part of a study on
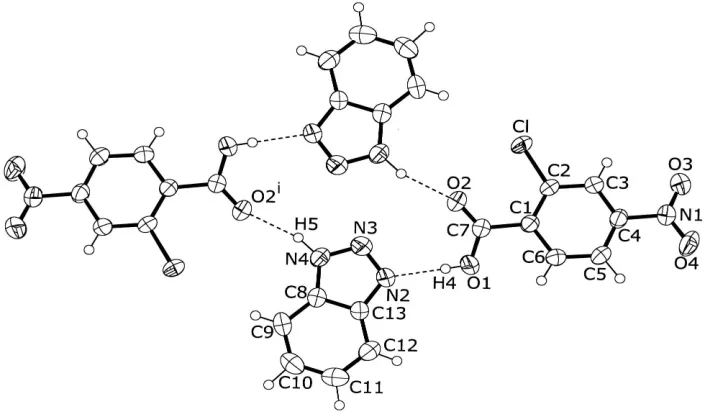
DÐH Ahydrogen bonding (D= N, O or C;A= N, O or Cl) in chloro-and nitro-substituted benzoic acid-amine systems (Ishidaet al., 2001a,b,c,d,e). In the crystal, two acid and two base components are held together by short OÐH N hydrogen bonds and relatively long NÐH O hydrogen bonds (Table 2) to afford a centrosymmetric macrocycle with graph-set descriptorR44(16) (Bernstein et al., 1995) (Fig. 1),
which is similar to that found in benzotriazole 3-nitrobenzoic acid (Hashizumeet al., 2001). The dihedral angle between the nitro group and the benzene ring is 10.03 (19), and that
between the carboxyl group and the benzene ring is 22.79 (17). A CÐH O hydrogen bond (C5ÐH2 O4ii;
Table 2) connects the hydrogen-bonded rings, resulting in the formation of a molecular ribbon running parallel to the [011] direction (Fig. 2). The ribbons, related by a twofold screw axis, are stacked along the a axis. A short contact [Cl O1iii,
3.164 (3) AÊ; symmetry code: (iii)1
2ÿx,12+y,32ÿz] is observed
between the ribbons.
Experimental
Crystals of (I) were obtained by slow evaporation from an aceto-nitrile solution of 1,2,3-benzotriazole and 2-chloro-4-nitrobenzoic acid in a molar ratio of 1:1.
Crystal data C7H4ClNO4C6H5N3 Mr= 320.69 Monoclinic,P21=n a= 7.0590 (15) AÊ
b= 11.7721 (13) AÊ
c= 16.4853 (17) AÊ
= 93.717 (13)
V= 1367.0 (4) AÊ3 Z= 4
Dx= 1.558 Mg mÿ3 Mo Kradiation Cell parameters from 25
re¯ections
= 10.5±12.5
= 0.30 mmÿ1 T= 300 K Prism, colorless 0.400.300.25 mm
Data collection
Rigaku AFC-5Rdiffractometer
!±2scans
Absorption correction: scan (Northet al., 1968)
Tmin= 0.913,Tmax= 0.927 3894 measured re¯ections 3134 independent re¯ections 1816 re¯ections withI> 2(I)
Rint= 0.020
max= 27.5 h=ÿ1!9
k= 0!15
l=ÿ21!21 3 standard re¯ections
every 97 re¯ections intensity decay: 1.4% Re®nement
Re®nement onF2 R[F2> 2(F2) = 0.052 wR(F2) = 0.132 S= 1.06 3134 re¯ections 236 parameters
All H-atom parameters re®ned
w= 1/[2(F
o2) + 0.6722P] whereP= (Fo2+ 2Fc2)/3 (/)max= 0.001
max= 0.25 e AÊÿ3
min=ÿ0.23 e AÊÿ3
Extinction correction:SHELXL
Extinction coef®cient: 4.7 (11)
10ÿ3 Table 1
Selected geometric parameters (AÊ). ClÐC2 1.723 (3) O1ÐC7 1.303 (4) O2ÐC7 1.196 (3) O3ÐN1 1.215 (3) O4ÐN1 1.214 (3) N1ÐC4 1.478 (3)
N2ÐN3 1.302 (3) N2ÐC13 1.379 (3) N3ÐN4 1.336 (3) N4ÐC8 1.360 (4) C1ÐC7 1.505 (4)
Table 2
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
O1ÐH4 N2 0.77 (5) 1.89 (5) 2.661 (3) 173 (5) N4ÐH5 O2i 0.95 (4) 2.00 (3) 2.909 (3) 158 (3) C5ÐH2 O4ii 0.97 (2) 2.48 (3) 3.265 (4) 138 (2)
Symmetry codes: (i) 1ÿx;1ÿy;2ÿz; (ii) 1ÿx;ÿy;1ÿz.
H atoms were found in difference Fourier maps and re®ned isotropically. Re®ned distances: CÐH = 0.89 (3)±1.03 (4), OÐH = 0.78 (4) and NÐH = 0.95 (4) AÊ.
Data collection: MSC/AFC Diffractometer Control Software
(Molecular Structure Corporation, 1990); cell re®nement:MSC/AFC Diffractometer Control Software; data reduction:teXsanfor Windows (Molecular Structure Corporation, 1997±1999); program(s) used to solve structure: SIR92 (Altomare et al.1993); program(s) used to re®ne structure:SHELXL97 (Sheldrick, 1997); molecular graphics:
ORTEP-3 (Farrugia, 1997); software used to prepare material for publication:teXsanfor Windows.
X-ray measurements were made at the X-ray Laboratory of Okayama University.
References
Altomare, A., Cascarano, G., Giacovazzo, C., & Guagliardi, A. (1993).J. Appl. Cryst.26, 343±350.
Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995).Angew. Chem. Int. Ed. Engl.34, 1555±1573.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Hashizume, D., Iegami, M., Yasui, M., Iwasaki, F., Meng, J., Wen, Z. & Matuura T. (2001).Acta Cryst.C57, 1067±1072.
Ishida, H., Rahman, B. & Kashino, S. (2001a).Acta Cryst.C57, 876±879. Ishida, H., Rahman, B. & Kashino, S. (2001b).Acta Cryst.C57, 1450±1453. Ishida, H., Rahman, B. & Kashino, S. (2001c).Acta Cryst.E57, o627±o629. Ishida, H., Rahman, B. & Kashino, S. (2001d).Acta Cryst.E57, o630±o632. Ishida, H., Rahman, B. & Kashino, S. (2001e).Acta Cryst.E57, o744±o745. Molecular Structure Corporation. (1990).MSC/AFC Diffractometer Control
Software. MSC, 3200 Research Forest Drive, The Woodlands, TX 77381, USA.
Molecular Structure Corporation. (1997±1999).teXsanfor Windows. Version 1.06. MSC, 9009 New Trails Drive, The Woodlands, TX 77381, USA. North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst.A24, 351±
359.
Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany.
Figure 1
ORTEP-3 (Farrugia, 1997) drawing of a hydrogen-bonded ring of (I), with the atom-labeling. Displacement ellipsoids of non-H atoms are drawn at the 50% probability level. OÐH N and NÐH O hydrogen bonds are indicated by dashed lines [symmetry code: (i) 1ÿx, 1ÿy, 2ÿz].
Figure 2
supporting information
sup-1
Acta Cryst. (2002). E58, o1083–o1084
supporting information
Acta Cryst. (2002). E58, o1083–o1084 [doi:10.1107/S1600536802015969]
The 1:1 complex of 2-chloro-4-nitrobenzoic acid and 1,2,3-benzotriazole
Hiroyuki Ishida, Takeo Fukunaga and Setsuo Kashino
S1. Comment
The title compound, (I), was investigated as part of a study on D—H···A hydrogen bonding (D = N, O or C; A = N, O or
Cl) in chloro-and nitro-substituted benzoic acid-amine systems (Ishida et al., 2001a,b,c,d,e). In the crystal, two acid and
two base components are held together by short O—H···N hydrogen bonds and relatively long N—H···O hydrogen bonds
(Table 2) to afford a centrosymmetric macro ring with graph-set descriptor R44(16) (Bernstein et al., 1995) (Fig. 1), which
is similar to that found in benzotriazole 3-nitrobenzoic acid (Hashizume et al., 2001). The dihedral angle between the
nitro group and the benzene ring is 10.03 (19)°, and that between the carboxyl group and the benzene ring is 22.79 (17)°.
A C—H···O hydrogen bond (C5—H2···O4ii; Table 2) connects the hydrogen-bonded rings, resulting in the formation of
molecular ribbon running parallel to the [011] direction (Fig. 2). The ribbons, related by a twofold screw axis, are stacked
along the a axis. A short contact [Cl···O1iii, 3.164 (3) Å; symmetry code: (iii) 1/2 − x, 1/2 + y, 3/2 − z] is observed
between the ribbons.
S2. Experimental
Crystals of (I) were obtained by slow evaporation from an acetonitrile solution of 1,2,3-benzotriazole with
2-chloro-4-nitrobenzoic acid with molar ratio of 1:1.
S3. Refinement
H atoms were found in difference Fourier maps and refined isotropically. Refined distances: C—H = 0.89 (3) − 1.03 (4),
Figure 1
ORTEP-3 (Farrugia, 1997) drawing of a hydrogen-bonded ring of (I), with the atom-labeling. Displacement ellipsoids of
non-H atoms are drawn at the 50% probability level. O—H···N and N—H···O hydrogen bonds are indicated by dashed
lines [symmetry code: (i) 1 − x, 1 − y, 2 − z].
Figure 2
Packing diagram, showing a molecular ribbon formed via C—H···O hydrogen bonds (indicated by dotted lines). O—
H···N and N—H···O hydrogen bonds are shown by dashed lines [symmetry codes are as in Table 2].
(I)
Crystal data
C7H4ClNO4·C6H5N3 Mr = 320.69
Monoclinic, P21/n Hall symbol: -P 2yn a = 7.0590 (15) Å b = 11.7721 (13) Å c = 16.4853 (17) Å
β = 93.717 (13)° V = 1367.0 (4) Å3 Z = 4
F(000) = 656.00 Dx = 1.558 Mg m−3
[image:4.610.129.481.355.538.2]supporting information
sup-3
Acta Cryst. (2002). E58, o1083–o1084
θ = 10.5–12.5° µ = 0.30 mm−1 T = 300 K
Prismatic, colorless 0.40 × 0.30 × 0.25 mm
Data collection
Rigaku AFC-5R diffractometer
Radiation source: Rigaku rotating anode Graphite monochromator
ω–2θ scans
Absorption correction: ψ scan (North et al., 1968)
Tmin = 0.913, Tmax = 0.927 3894 measured reflections
3134 independent reflections 1816 reflections with I > 2σ(I) Rint = 0.020
θmax = 27.5°, θmin = 2.1° h = −1→9
k = 0→15 l = −21→21
3 standard reflections every 97 reflections intensity decay: 1.4%
Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.052 wR(F2) = 0.132 S = 1.06 3134 reflections 236 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
All H-atom parameters refined w = 1/[σ2(F
o2) + 0.6722P] where P = (Fo2 + 2Fc2)/3 (Δ/σ)max = 0.001
Δρmax = 0.25 e Å−3 Δρmin = −0.23 e Å−3
Extinction correction: SHELXL, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 Extinction coefficient: 4.7 (11)×10-3
Special details
Experimental. The scan width was (1.73 + 0.30tanθ)° with an ω scan speed of 6° per minute (up to 3 scans to achieve I/σ(I) > 10). Stationary background counts were recorded at each end of the scan, and the scan time:background time ratio was 2:1.
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Cl 0.38055 (15) 0.51916 (6) 0.64808 (5) 0.0581 (3)
O1 0.3549 (4) 0.2435 (2) 0.82590 (14) 0.0724 (9)
O2 0.4708 (4) 0.41516 (18) 0.80647 (13) 0.0680 (8)
O3 0.3291 (5) 0.2687 (2) 0.39476 (13) 0.0903 (11)
O4 0.3893 (4) 0.09565 (19) 0.42854 (13) 0.0726 (8)
N1 0.3657 (4) 0.1950 (2) 0.44490 (15) 0.0514 (7)
N2 0.3769 (4) 0.2854 (2) 0.98486 (14) 0.0533 (7)
N4 0.4115 (4) 0.3974 (2) 1.08682 (16) 0.0530 (8)
C1 0.3985 (4) 0.2946 (2) 0.69216 (16) 0.0369 (7)
C2 0.3887 (4) 0.3751 (2) 0.63032 (16) 0.0375 (7)
C3 0.3785 (5) 0.3416 (2) 0.54951 (17) 0.0409 (7)
C4 0.3780 (4) 0.2282 (2) 0.53154 (16) 0.0397 (7)
C5 0.3874 (5) 0.1449 (2) 0.59007 (18) 0.0441 (7)
C6 0.3971 (5) 0.1798 (2) 0.67076 (18) 0.0442 (8)
C7 0.4109 (5) 0.3263 (2) 0.78078 (16) 0.0434 (7)
C8 0.4035 (4) 0.2920 (2) 1.12031 (16) 0.0401 (7)
C9 0.4128 (5) 0.2515 (3) 1.20022 (18) 0.0516 (9)
C10 0.4004 (5) 0.1374 (3) 1.2092 (2) 0.0565 (9)
C11 0.3807 (5) 0.0632 (3) 1.1426 (2) 0.0591 (9)
C12 0.3692 (5) 0.1032 (3) 1.0646 (2) 0.0516 (9)
C13 0.3811 (4) 0.2195 (2) 1.05411 (16) 0.0388 (7)
H1 0.368 (4) 0.392 (2) 0.5099 (15) 0.033 (7)*
H2 0.385 (4) 0.066 (2) 0.5739 (16) 0.047 (8)*
H3 0.408 (4) 0.130 (2) 0.7132 (16) 0.037 (8)*
H4 0.370 (7) 0.254 (4) 0.872 (3) 0.109 (17)*
H5 0.428 (6) 0.470 (3) 1.112 (2) 0.096 (14)*
H6 0.433 (5) 0.298 (3) 1.2423 (19) 0.061 (10)*
H7 0.407 (5) 0.102 (3) 1.266 (2) 0.088 (12)*
H8 0.373 (4) −0.015 (3) 1.1547 (17) 0.047 (8)*
H9 0.360 (5) 0.052 (3) 1.023 (2) 0.072 (11)*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-5
Acta Cryst. (2002). E58, o1083–o1084
C13 0.0431 (18) 0.0399 (15) 0.0331 (13) −0.0018 (14) 0.0013 (13) −0.0022 (11)
Geometric parameters (Å, º)
Cl—C2 1.723 (3) C3—C4 1.367 (4)
O1—C7 1.303 (4) C3—H1 0.89 (3)
O1—H4 0.78 (4) C4—C5 1.374 (4)
O2—C7 1.196 (3) C5—C6 1.390 (4)
O3—N1 1.215 (3) C5—H2 0.97 (3)
O4—N1 1.214 (3) C6—H3 0.91 (3)
N1—C4 1.478 (3) C8—C13 1.387 (4)
N2—N3 1.302 (3) C8—C9 1.398 (4)
N2—C13 1.379 (3) C9—C10 1.356 (5)
N3—N4 1.336 (3) C9—H6 0.89 (3)
N4—C8 1.360 (4) C10—C11 1.403 (5)
N4—H5 0.95 (4) C10—H7 1.03 (4)
C1—C2 1.391 (4) C11—C12 1.368 (5)
C1—C6 1.396 (4) C11—H8 0.95 (3)
C1—C7 1.505 (4) C12—C13 1.383 (4)
C2—C3 1.387 (4) C12—H9 0.91 (3)
Cl···O1i 3.164 (3) O4···C5vi 3.265 (4)
Cl···O3ii 3.335 (3) N2···C7 3.425 (4)
O1···N2 2.661 (3) N3···N3iii 2.962 (4)
O2···N4iii 2.909 (3) N3···N4iii 3.260 (4)
O3···C9iv 3.305 (4) N4···C5vii 3.393 (5)
O4···N4v 3.154 (4) C1···C9viii 3.482 (5)
C7—O1—H4 115 (3) C5—C6—C1 121.8 (3)
O4—N1—O3 124.3 (3) C5—C6—H3 122.5 (16)
O4—N1—C4 118.0 (2) C1—C6—H3 115.6 (16)
O3—N1—C4 117.7 (2) O2—C7—O1 124.5 (3)
N3—N2—C13 109.0 (2) O2—C7—C1 123.9 (3)
N2—N3—N4 108.3 (2) O1—C7—C1 111.6 (2)
N3—N4—C8 110.9 (2) N4—C8—C13 104.3 (2)
N3—N4—H5 118 (2) N4—C8—C9 133.8 (3)
C8—N4—H5 131 (2) C13—C8—C9 121.8 (3)
C2—C1—C6 118.4 (2) C10—C9—C8 116.2 (3)
C2—C1—C7 122.6 (2) C10—C9—H6 122 (2)
C6—C1—C7 119.0 (2) C8—C9—H6 122 (2)
C3—C2—C1 120.5 (2) C9—C10—C11 122.4 (3)
C3—C2—Cl 116.3 (2) C9—C10—H7 120 (2)
C1—C2—Cl 123.2 (2) C11—C10—H7 117 (2)
C4—C3—C2 119.0 (3) C12—C11—C10 121.2 (3)
C4—C3—H1 120.1 (17) C12—C11—H8 122.2 (18)
C2—C3—H1 120.9 (17) C10—C11—H8 116.5 (18)
C3—C4—C5 123.0 (3) C11—C12—C13 117.2 (3)
C5—C4—N1 119.1 (2) C13—C12—H9 124 (2)
C4—C5—C6 117.2 (3) N2—C13—C12 131.4 (3)
C4—C5—H2 119.6 (17) N2—C13—C8 107.5 (2)
C6—C5—H2 123.2 (17) C12—C13—C8 121.1 (3)
C13—N2—N3—N4 0.0 (4) C2—C1—C7—O2 −24.0 (5)
N2—N3—N4—C8 −0.1 (4) C6—C1—C7—O2 155.9 (3)
C6—C1—C2—C3 −0.2 (5) C2—C1—C7—O1 158.3 (3)
C7—C1—C2—C3 179.6 (3) C6—C1—C7—O1 −21.8 (4)
C6—C1—C2—Cl 177.6 (2) N3—N4—C8—C13 0.1 (4)
C7—C1—C2—Cl −2.6 (4) N3—N4—C8—C9 −179.8 (4)
C1—C2—C3—C4 0.0 (5) N4—C8—C9—C10 −179.5 (4)
Cl—C2—C3—C4 −177.9 (2) C13—C8—C9—C10 0.6 (5)
C2—C3—C4—C5 −0.1 (5) C8—C9—C10—C11 0.4 (6)
C2—C3—C4—N1 179.6 (3) C9—C10—C11—C12 −1.3 (6)
O4—N1—C4—C3 170.9 (3) C10—C11—C12—C13 1.1 (6)
O3—N1—C4—C3 −10.5 (5) N3—N2—C13—C12 −179.3 (4)
O4—N1—C4—C5 −9.5 (5) N3—N2—C13—C8 0.0 (4)
O3—N1—C4—C5 169.2 (3) C11—C12—C13—N2 179.1 (3)
C3—C4—C5—C6 0.2 (5) C11—C12—C13—C8 −0.1 (5)
N1—C4—C5—C6 −179.4 (3) N4—C8—C13—N2 0.0 (4)
C4—C5—C6—C1 −0.4 (5) C9—C8—C13—N2 179.9 (3)
C2—C1—C6—C5 0.4 (5) N4—C8—C13—C12 179.3 (3)
C7—C1—C6—C5 −179.4 (3) C9—C8—C13—C12 −0.7 (5)
Symmetry codes: (i) −x+1/2, y+1/2, −z+3/2; (ii) −x+1, −y+1, −z+1; (iii) −x+1, −y+1, −z+2; (iv) x, y, z−1; (v) −x+1/2, y−1/2, −z+3/2; (vi) −x+1, −y, −z+1; (vii) x+1/2, −y+1/2, z+1/2; (viii) x−1/2, −y+1/2, z−1/2.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O1—H4···N2 0.77 (5) 1.89 (5) 2.661 (3) 173 (5)
N4—H5···O2iii 0.95 (4) 2.00 (3) 2.909 (3) 158 (3)
C5—H2···O4vi 0.97 (2) 2.48 (3) 3.265 (4) 138 (2)