organic papers
o680
Fuet al. C15H12O4 doi:10.1107/S1600536806001760 Acta Cryst.(2006). E62, o680–o681
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
Methyl phenyl terephthalate
Xian-Shu Fu,aFa Chengb* and Yong-Mei Xiec
a
College of Life Sciences, China Jiliang University, Hangzhou 310018, People’s Republic of China,bDepartment of Chemistry, Tianjin University, Tianjin 300072, People’s Republic of China, andcDepartment of Chemical Engineering, Hangzhou Vocational Technology Institute, Hangzhou 310018, People’s Republic of China
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 294 K
Mean(C–C) = 0.003 A˚
Rfactor = 0.043
wRfactor = 0.117
Data-to-parameter ratio = 14.5
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 21 December 2005 Accepted 16 January 2006
#2006 International Union of Crystallography
All rights reserved
The title compound, C15H12O4, was synthesized in an
anhydrous medium. The aromatic rings make a dihedral angle of 37.43 (5). Weak intermolecular C—H O hydrogen bonds involving one of the carbonyl O atoms stabilize the crystal packing.
Comment
Organic electroluminescence – the emission of light by organic molecules exposed to an electric field – has been extensively investigated in academic and industrial laboratories (Cui & Kim, 2004). The title compound, (I) (Fig. 1), prepared by our group, belongs to the family of organic electroluminescent materials. We report here its X-ray crystal structure.
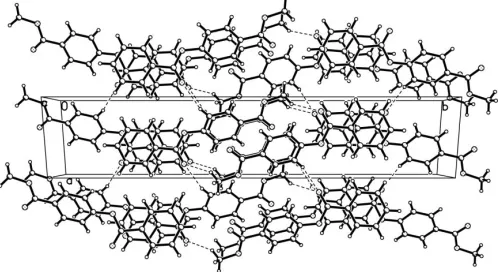
The bond lengths and angles in (I) (Table 1) show normal values. The two aromatic rings make a dihedral angle of 37.43 (5). Weak intermolecular C—H O hydrogen bonds (Table 2) stabilize the crystal packing (Fig. 2).
Experimental
The title compound was prepared according to a known procedure (Li et al., 2000), which resulted in a colourless powder (m.p. 405– 406 K). Single crystals suitable for X-ray analysis were obtained by slow evaporation of a dichloromethane solution.
Crystal data C15H12O4
Mr= 256.25
Monoclinic,P21=c
a= 6.1654 (13) A˚
b= 29.374 (6) A˚
c= 7.2827 (16) A˚
= 111.188 (4)
V= 1229.8 (5) A˚3
Z= 4
Dx= 1.384 Mg m
3 MoKradiation Cell parameters from 2124
reflections
= 2.8–25.8 = 0.10 mm1
T= 294 (2) K Block, colourless 0.300.280.20 mm
Data collection
Bruker SMART CCD area-detector diffractometer
’and!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 2002)
Tmin= 0.967,Tmax= 0.980 6896 measured reflections
2526 independent reflections 1623 reflections withI> 2(I)
Rint= 0.047
max= 26.5
h=3!7
k=36!36
Refinement Refinement onF2 R[F2> 2(F2)] = 0.043
wR(F2) = 0.117
S= 1.01 2526 reflections 174 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.0459P)2 + 0.2662P]
whereP= (Fo2+ 2Fc2)/3 (/)max= 0.001
max= 0.16 e A˚
3
min=0.19 e A˚
3
Extinction correction:SHELXL97
[image:2.610.316.565.74.210.2]Extinction coefficient: 0.018 (2)
Table 1
Selected geometric parameters (A˚ ,).
O1—C7 1.194 (2) O2—C7 1.352 (2)
O2—C8 1.401 (2) C6—C7 1.478 (2)
C5—C6—C7 118.13 (16) O1—C7—C6 125.13 (16)
C9—C8—O2 123.42 (16)
C8—O2—C7—C6 177.84 (15) C5—C6—C7—O1 6.9 (3)
C5—C6—C7—O2 174.25 (15) O2—C8—C9—C10 173.84 (16)
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
C13—H13 O1i
0.93 2.46 3.362 (2) 164 C15—H15A O1ii
0.96 2.57 3.443 (2) 152
Symmetry codes: (i)x1;y;z; (ii)xþ1;yþ1;zþ2.
All H atoms were positioned geometrically and refined as riding (C—H = 0.93 or 0.96 A˚ ). For CH groups, Uiso(H) values were set
equal to 1.2Ueq(C) and for the methyl groups they were set equal to
1.5Ueq(C).
Data collection:SMART(Bruker, 1997); cell refinement:SAINT
(Bruker, 1997); data reduction:SAINT; program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
SHELXTL (Bruker, 1997); software used to prepare material for publication:SHELXTL.
This work was supported by the Scientific Research Foundation for Returned Overseas Chinese Scholars and the Ministry of Education.
References
Bruker (1997).SMART,SAINTandSHELXTL(Version 5.10). Bruker AXS Inc, Madison, Wiscosin, USA.
Cui, J. Z. & Kim, S. H. (2004).Chin. Sci. Bull.49, 797–802. Li, W. R., Yo, Y. C. & Lin, Y. S. (2000).Tetrahedron,56, 8867–8875. Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of
Go¨ttingen, Germany.
[image:2.610.46.299.74.158.2]Sheldrick, G. M. (2002).SADABS. Version 2.03. University of Go¨ttingen, Germany.
Figure 1
View of (I), showing the atom-numbering scheme and displacement ellipsoids drawn at the 35% probability level.
Figure 2
[image:2.610.44.297.362.436.2]supporting information
sup-1 Acta Cryst. (2006). E62, o680–o681
supporting information
Acta Cryst. (2006). E62, o680–o681 [https://doi.org/10.1107/S1600536806001760]
Methyl phenyl terephthalate
Xian-Shu Fu, Fa Cheng and Yong-Mei Xie
Methyl phenyl terephthalate
Crystal data
C15H12O4 Mr = 256.25 Monoclinic, P21/c a = 6.1654 (13) Å
b = 29.374 (6) Å
c = 7.2827 (16) Å
β = 111.188 (4)°
V = 1229.8 (5) Å3 Z = 4
F(000) = 536
Dx = 1.384 Mg m−3
Melting point: 405 K
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2124 reflections
θ = 2.8–25.8°
µ = 0.10 mm−1 T = 294 K Block, colourless 0.30 × 0.28 × 0.20 mm
Data collection
Bruker SMART CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 2002)
Tmin = 0.967, Tmax = 0.980
6896 measured reflections 2526 independent reflections 1623 reflections with I > 2σ(I)
Rint = 0.047
θmax = 26.5°, θmin = 1.4° h = −3→7
k = −36→36
l = −9→9
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.043 wR(F2) = 0.117 S = 1.01 2526 reflections 174 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0459P)2 + 0.2662P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.16 e Å−3
Δρmin = −0.19 e Å−3
Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full
covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.8236 (2) 0.33457 (4) 0.9330 (2) 0.0562 (4)
O2 0.4342 (2) 0.33467 (4) 0.79063 (19) 0.0470 (4)
O3 0.3580 (3) 0.55076 (4) 0.6906 (2) 0.0640 (4)
O4 0.0482 (2) 0.53167 (4) 0.76334 (19) 0.0507 (4)
C1 0.4101 (3) 0.24240 (6) 0.7940 (3) 0.0501 (5)
H1 0.2740 0.2594 0.7591 0.060*
C2 0.3988 (4) 0.19530 (7) 0.7851 (3) 0.0624 (6)
H2 0.2552 0.1807 0.7423 0.075*
C3 0.5987 (4) 0.17031 (7) 0.8392 (3) 0.0617 (6)
H3 0.5910 0.1387 0.8334 0.074*
C4 0.8102 (4) 0.19157 (7) 0.9018 (3) 0.0644 (6)
H4 0.9460 0.1744 0.9404 0.077*
C5 0.8226 (3) 0.23852 (7) 0.9080 (3) 0.0525 (5)
H5 0.9666 0.2529 0.9487 0.063*
C6 0.6222 (3) 0.26409 (6) 0.8543 (2) 0.0368 (4)
C7 0.6446 (3) 0.31421 (6) 0.8650 (3) 0.0383 (4)
C8 0.4107 (3) 0.38211 (6) 0.7909 (3) 0.0395 (4)
C9 0.5444 (3) 0.41156 (6) 0.7299 (3) 0.0445 (5)
H9 0.6676 0.4009 0.6971 0.053*
C10 0.4916 (3) 0.45736 (6) 0.7184 (3) 0.0447 (5)
H10 0.5821 0.4778 0.6796 0.054*
C11 0.3060 (3) 0.47338 (6) 0.7637 (2) 0.0376 (4)
C12 0.1765 (3) 0.44305 (6) 0.8270 (3) 0.0454 (5)
H12 0.0534 0.4536 0.8605 0.054*
C13 0.2293 (3) 0.39713 (6) 0.8407 (3) 0.0454 (5)
H13 0.1424 0.3767 0.8834 0.055*
C14 0.2462 (3) 0.52242 (6) 0.7359 (3) 0.0421 (4)
C15 −0.0304 (4) 0.57823 (6) 0.7333 (3) 0.0553 (5)
H15A 0.0782 0.5972 0.8309 0.083*
H15B −0.1805 0.5805 0.7443 0.083*
H15C −0.0412 0.5880 0.6045 0.083*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3 Acta Cryst. (2006). E62, o680–o681
O2 0.0392 (7) 0.0302 (7) 0.0666 (9) 0.0000 (5) 0.0131 (6) −0.0001 (6) O3 0.0714 (10) 0.0365 (8) 0.0958 (12) −0.0030 (7) 0.0443 (9) 0.0113 (7) O4 0.0544 (8) 0.0328 (7) 0.0687 (9) 0.0050 (6) 0.0268 (7) 0.0051 (6) C1 0.0433 (11) 0.0388 (10) 0.0640 (13) −0.0016 (9) 0.0145 (10) −0.0020 (9) C2 0.0635 (14) 0.0409 (11) 0.0814 (16) −0.0127 (11) 0.0244 (12) −0.0062 (11) C3 0.0898 (18) 0.0318 (11) 0.0681 (14) 0.0013 (11) 0.0339 (13) −0.0018 (10) C4 0.0671 (15) 0.0441 (12) 0.0778 (16) 0.0166 (11) 0.0212 (13) 0.0028 (11) C5 0.0462 (11) 0.0465 (12) 0.0612 (13) 0.0027 (9) 0.0150 (10) −0.0009 (10) C6 0.0428 (10) 0.0329 (9) 0.0347 (9) 0.0010 (8) 0.0139 (8) −0.0005 (7) C7 0.0375 (10) 0.0369 (10) 0.0394 (10) −0.0013 (8) 0.0128 (8) 0.0000 (8) C8 0.0378 (10) 0.0302 (9) 0.0468 (10) −0.0020 (8) 0.0109 (8) 0.0014 (8) C9 0.0428 (10) 0.0386 (10) 0.0558 (12) 0.0021 (8) 0.0224 (9) 0.0027 (8) C10 0.0456 (11) 0.0372 (10) 0.0537 (12) −0.0036 (9) 0.0209 (9) 0.0060 (8) C11 0.0398 (10) 0.0328 (9) 0.0395 (9) −0.0017 (8) 0.0136 (8) 0.0021 (7) C12 0.0443 (11) 0.0385 (10) 0.0589 (12) 0.0035 (8) 0.0253 (10) 0.0055 (9) C13 0.0438 (11) 0.0361 (10) 0.0603 (12) −0.0028 (8) 0.0236 (10) 0.0080 (8) C14 0.0477 (11) 0.0344 (9) 0.0431 (10) −0.0026 (8) 0.0151 (9) −0.0007 (8) C15 0.0664 (13) 0.0352 (10) 0.0630 (13) 0.0093 (10) 0.0219 (11) 0.0005 (9)
Geometric parameters (Å, º)
O1—C7 1.194 (2) C5—H5 0.9300
O2—C7 1.352 (2) C6—C7 1.478 (2)
O2—C8 1.401 (2) C8—C13 1.367 (2)
O3—C14 1.200 (2) C8—C9 1.375 (2)
O4—C14 1.335 (2) C9—C10 1.380 (2)
O4—C15 1.441 (2) C9—H9 0.9300
C1—C6 1.376 (2) C10—C11 1.383 (3)
C1—C2 1.386 (3) C10—H10 0.9300
C1—H1 0.9300 C11—C12 1.382 (2)
C2—C3 1.364 (3) C11—C14 1.482 (2)
C2—H2 0.9300 C12—C13 1.383 (2)
C3—C4 1.367 (3) C12—H12 0.9300
C3—H3 0.9300 C13—H13 0.9300
C4—C5 1.381 (3) C15—H15A 0.9600
C4—H4 0.9300 C15—H15B 0.9600
C5—C6 1.376 (2) C15—H15C 0.9600
C7—O2—C8 121.76 (13) C9—C8—O2 123.42 (16)
C14—O4—C15 116.49 (15) C8—C9—C10 118.51 (17)
C6—C1—C2 120.22 (18) C8—C9—H9 120.7
C6—C1—H1 119.9 C10—C9—H9 120.7
C2—C1—H1 119.9 C9—C10—C11 120.99 (17)
C3—C2—C1 119.91 (19) C9—C10—H10 119.5
C3—C2—H2 120.0 C11—C10—H10 119.5
C1—C2—H2 120.0 C12—C11—C10 119.20 (16)
C2—C3—C4 120.24 (19) C12—C11—C14 121.81 (17)
C4—C3—H3 119.9 C11—C12—C13 120.24 (17)
C3—C4—C5 120.1 (2) C11—C12—H12 119.9
C3—C4—H4 119.9 C13—C12—H12 119.9
C5—C4—H4 119.9 C8—C13—C12 119.31 (17)
C6—C5—C4 120.16 (19) C8—C13—H13 120.3
C6—C5—H5 119.9 C12—C13—H13 120.3
C4—C5—H5 119.9 O3—C14—O4 123.15 (17)
C1—C6—C5 119.32 (16) O3—C14—C11 124.93 (18)
C1—C6—C7 122.55 (16) O4—C14—C11 111.90 (16)
C5—C6—C7 118.13 (16) O4—C15—H15A 109.5
O1—C7—O2 123.53 (15) O4—C15—H15B 109.5
O1—C7—C6 125.13 (16) H15A—C15—H15B 109.5
O2—C7—C6 111.33 (15) O4—C15—H15C 109.5
C13—C8—C9 121.72 (16) H15A—C15—H15C 109.5
C13—C8—O2 114.62 (15) H15B—C15—H15C 109.5
C6—C1—C2—C3 1.0 (3) C13—C8—C9—C10 −0.3 (3)
C1—C2—C3—C4 −0.1 (3) O2—C8—C9—C10 173.84 (16)
C2—C3—C4—C5 −0.9 (3) C8—C9—C10—C11 −1.1 (3)
C3—C4—C5—C6 1.1 (3) C9—C10—C11—C12 1.9 (3)
C2—C1—C6—C5 −0.9 (3) C9—C10—C11—C14 −175.79 (17)
C2—C1—C6—C7 179.50 (19) C10—C11—C12—C13 −1.3 (3)
C4—C5—C6—C1 −0.2 (3) C14—C11—C12—C13 176.33 (17)
C4—C5—C6—C7 179.49 (19) C9—C8—C13—C12 0.9 (3)
C8—O2—C7—O1 −1.1 (3) O2—C8—C13—C12 −173.73 (16)
C8—O2—C7—C6 177.84 (15) C11—C12—C13—C8 0.0 (3)
C1—C6—C7—O1 172.78 (18) C15—O4—C14—O3 0.8 (3)
C5—C6—C7—O1 −6.9 (3) C15—O4—C14—C11 −177.97 (15)
C1—C6—C7—O2 −6.1 (2) C12—C11—C14—O3 176.19 (19)
C5—C6—C7—O2 174.25 (15) C10—C11—C14—O3 −6.1 (3)
C7—O2—C8—C13 −138.14 (17) C12—C11—C14—O4 −5.1 (2)
C7—O2—C8—C9 47.4 (3) C10—C11—C14—O4 172.63 (16)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C13—H13···O1i 0.93 2.46 3.362 (2) 164
C15—H15A···O1ii 0.96 2.57 3.443 (2) 152