ISOCHROMOSOME Xq : NOT A RARE FINDING IN SHORT STATURE FEMALES WITH
*Sanchita Roy, Ajanta Haldar, Pritha Pal, Atreyee
Institute of Post
ARTICLE INFO ABSTRACT
Iso-chromosome of long arm of X chromosome i(Xq
cleavage of the centromere during cell division, found in some cases Turner Syndrome. The aim of this study is to estimate the prevalence of isochromosome X in patients with short stature with or with-
done comprised of female patients who had been referred to our cytogenetic outdoor of Vivekananda Institute of Medical Sciences (VIMS), Kolkata, India with the history o
amenorrhoea between 2012, January patients aged between 10
amenorrhea who were referred to th
departments of Pediatric medicine, Endocrinology, Gynaecology from different hospitals of Eastern India were included in the study. Lymphocyte culture depended karyo typing done using conventiona
patients presented mainly with short stature with or without primary or secondary amenorrhea. Our study re
without primary or secondary amenorrhea to remove the diagnostic dilemma of the clinician.
Copyright © 2015 Sanchita Roy et al. This is an open access article distributed under the Creative Commons Att distribution, and reproduction in any medium, provided the original work is properly cited.
INTRODUCTION
Short stature is defined as a standing height more than 2 standard deviations (SDs) below the mean (or below the 2.5 percentile) for sex according to standard growth chart
et al., 2007) and is believed to have a strong genetic component. The genetic abnormalities may lie in sex chromosomes as well as in autosomes. Turner syndrome (TS) is considered to be the important genetic abnormalities leading to short stature in females affecting 1 in every 2000 girls. (Donaldson et al., 2006) TS is characterized cytogenetically either by X chromosome monosomy(45,X),
abnormal X chromosome(isochromosome/ring chromosome/ deletion in short arm), or mosaicism of a 45,
another cell line, which might be 46,XX, 46,XY or have an abnormal sex chromosome rearrangement like isochromosome or ring chromosome etc. (Jacobs et al., 1997
analysis, the percentage occurrences of the various karyotypes observed in TS are documented in different studies.
et al., 2007; Sybert and McCauley, 2004) The 45 X cell line arises from meiotic non-disjunction or anaphase lagging during spermatogenesis or oogenesis or from post zygotic error.
*Corresponding author: Sanchita Roy,
Institute of Post–Graduation Medical Education and Research
ISSN: 0975-833X
Article History:
Received 14th March, 2015
Received in revised form 06th April, 2015 Accepted 14th May, 2015
Published online 27th June,2015
Key words:
Isochromosome, Turner syndrome, Karyotyping,
Primary and secondary amenorrhea, Short stature.
Citation:Sanchita Roy, Ajanta Haldar, Pritha Pal, Atreyee Dutta
stature females with amenorrhoea”, International Journal of Current Research
RESEARCH ARTICLE
: NOT A RARE FINDING IN SHORT STATURE FEMALES WITH
AMENORRHOEA
*Sanchita Roy, Ajanta Haldar, Pritha Pal, Atreyee Dutta and Shanoli Ghosh
Institute of Post–Graduation Medical Education and Research
ABSTRACT
chromosome of long arm of X chromosome i(Xq), which occurs due to abnormal transverse cleavage of the centromere during cell division, found in some cases Turner Syndrome. The aim of this study is to estimate the prevalence of isochromosome X in patients with short stature with or out primary (PA) and secondary amenorrhea (SA) in our population. An observational study done comprised of female patients who had been referred to our cytogenetic outdoor of Vivekananda Institute of Medical Sciences (VIMS), Kolkata, India with the history o
amenorrhoea between 2012, January-2014, December and designed as original article. The female patients aged between 10-25years having short stature with or with
amenorrhea who were referred to the cytogenetic outdoor for chromosomal analysis from the departments of Pediatric medicine, Endocrinology, Gynaecology from different hospitals of Eastern India were included in the study. Lymphocyte culture depended karyo typing done using conventional method. We have documented seven such cases of is chromosome Xq among 50 female patients presented mainly with short stature with or without primary or secondary amenorrhea. Our study re-emphasizes the importance of chromosomal analysis in short statu
without primary or secondary amenorrhea to remove the diagnostic dilemma of the clinician.
is an open access article distributed under the Creative Commons Attribution License, which distribution, and reproduction in any medium, provided the original work is properly cited.
Short stature is defined as a standing height more than 2 standard deviations (SDs) below the mean (or below the 2.5 according to standard growth chart (Cohen and is believed to have a strong genetic component. The genetic abnormalities may lie in sex chromosomes as well as in autosomes. Turner syndrome (TS) is considered to be the important genetic abnormalities leading in every 2000 girls. TS is characterized cytogenetically the presence of an abnormal X chromosome(isochromosome/ring chromosome/ in short arm), or mosaicism of a 45, X cell line with another cell line, which might be 46,XX, 46,XY or have an abnormal sex chromosome rearrangement like isochromosome 1997) On chromosomal analysis, the percentage occurrences of the various karyotypes observed in TS are documented in different studies. (Graham
The 45 X cell line disjunction or anaphase lagging during spermatogenesis or oogenesis or from post zygotic error.
Graduation Medical Education and Research
Iso-chromosome is defined as the structurally abnormal chromosome consisting of either two short or long arms, because of the abnormal transverse misdivision
centromere resulting in unbalanced chromosomal constitution, monosomy for the missing and trisomy for the duplicated arms. The process of isochromosome may occur in premeiotic gamete, during meiotic cell divisions or in post zygotic cell divisions of a normal or trisomic conceptus
manifestations of classical TS usually include short stature, webbed neck, broad chest with widely spaced nipples, cubitus valgus, congenital lymphedema, lack of spontaneous pubertal development resulting from ovarian sex hormone insufficiency, primary (PA) or secondary amenorrhea
hairline, misshapen or rotated ears, narrow palate with crowded teeth, hyper-convex nails, multi
malformation6 but the X iso
present with short stature with absence or minor Turner stigmata thus creating really a challenge to the clinicians Absence of typical phenotypic feature of TS will cause delay in diagnosis as well as delayed treatment with hormonal supplementation. Treatment with recombinant human growth hormone increases height velocity and ultimate stature in most, but the best results of growth hormone treatment have been found if it is initiated at age 4-5 years.
International Journal of Current Research
Vol. 7, Issue, 06, pp.16876-16880, June, 2015
INTERNATIONAL
Sanchita Roy, Ajanta Haldar, Pritha Pal, Atreyee Dutta and Shanoli Ghosh, 2015. “Isochromosome Xq : International Journal of Current Research, 7, (6), 16876-16880.
: NOT A RARE FINDING IN SHORT STATURE FEMALES WITH
Shanoli Ghosh
Graduation Medical Education and Research
), which occurs due to abnormal transverse cleavage of the centromere during cell division, found in some cases Turner Syndrome. The aim of this study is to estimate the prevalence of isochromosome X in patients with short stature with or rimary (PA) and secondary amenorrhea (SA) in our population. An observational study done comprised of female patients who had been referred to our cytogenetic outdoor of Vivekananda Institute of Medical Sciences (VIMS), Kolkata, India with the history of short stature with or without 2014, December and designed as original article. The female 25years having short stature with or with-out primary or secondary e cytogenetic outdoor for chromosomal analysis from the departments of Pediatric medicine, Endocrinology, Gynaecology from different hospitals of Eastern India were included in the study. Lymphocyte culture depended karyo typing done using l method. We have documented seven such cases of is chromosome Xq among 50 female patients presented mainly with short stature with or without primary or secondary amenorrhea. Our emphasizes the importance of chromosomal analysis in short stature female patients with or without primary or secondary amenorrhea to remove the diagnostic dilemma of the clinician.
ribution License, which permits unrestricted use,
chromosome is defined as the structurally abnormal chromosome consisting of either two short or long arms, because of the abnormal transverse misdivision of the centromere resulting in unbalanced chromosomal constitution, monosomy for the missing and trisomy for the duplicated arms. The process of isochromosome may occur in premeiotic gamete, during meiotic cell divisions or in post zygotic cell a normal or trisomic conceptus. (Fig. 1) Clinical manifestations of classical TS usually include short stature, webbed neck, broad chest with widely spaced nipples, cubitus valgus, congenital lymphedema, lack of spontaneous pubertal ting from ovarian sex hormone insufficiency, (PA) or secondary amenorrhea (SA), a low-posterior hairline, misshapen or rotated ears, narrow palate with crowded convex nails, multi-pigmented nevi, and cardiac so-chromosome patients mainly present with short stature with absence or minor Turner y a challenge to the clinicians. Absence of typical phenotypic feature of TS will cause delay in diagnosis as well as delayed treatment with hormonal Treatment with recombinant human growth hormone increases height velocity and ultimate stature in most, ts of growth hormone treatment have been
5 years. (Lanes, 2004)
INTERNATIONAL JOURNAL OF CURRENT RESEARCH
Therefore any female with short stature with or without PA or SA must alert the clinician to the possibility of this variant type of Turners Syndrome.
[image:2.595.39.290.350.742.2]Fig. 1. Formation of isochromosome due to abnormal centromeric division
Fig. 2. Karyotype of monosomy X, 45 X
Fig. 3. Karyotype of isochromosome X of long arm, 46 XXi(q10)
This study is done to find out the prevalence of iso-chromosome X in female patients with short stature in our population of West-Bengal of India which as per of our knowledge have not been reported previously.
MATERIALS AND METHODS
This study was carried out in the Department of Genetics in Vivekananda Institute of Medical Sciences (VIMS), Kolkata, West Bengal during the period Jan 2012 - Dec 2014 and approved by the ethical committee of VIMS. Informed consent was obtained from the patients after the procedure was fully explained to them. The female patients having short stature with or with-out primary or secondary amenorrhea in the age group of 10-25 years were referred to the cytogenetic outdoor for chromosomal analysis from the departments of Pediatric medicine, Endocrinology, Gynaecology from different hospitals of West-Bengal.. A total of 50 patients were included in the study. The physical examination included the accurate measurement of the height and weight and thorough inspection and palpation of the external genitalia, the primary and secondary sexual characters and a search for the other congenital anomalies. Tanner-Whitehouse growth charts were used for monitoring the growth and assessment of heights among the patients. After taking brief clinical and family history, patients were advised for plasma hormonal levels (FSH, LH, Thyroid profile, oestradiol, serum growth hormone level, Insulin like growth hormone level) and blood karyotyping. The blood sample was collected from the patients in a completely sterile heparinised vacutainer tube and mixed well. The cultures were set up with RPMI 1640 (Rosewell Park Memorial Institute) culture medium. Peripherial blood lymphocytes inducted with 2% phytohaemagglutinin (PHA) were incubated at 37.5°C for 72 h. One and a half hours prior to harvest, the cultures were arrested with colchicine and treated with 0.75 M KCl (potassium chloride) for 30 min and fixed in 3:1 ratio of methanol/glacial acetic acid fixative. After air drying, conventional Giemsa (GTG) banding was performed to identify the chromosomes. After banding, 50 metaphases were scanned under low power for each case on OLYMPUS BX51 microscope and then 10 metaphases were analysed by automated karyotyping system (CYTOVISION software). In cases of mosaics 30 metaphases were analysed.
RESULTS
[image:2.595.46.284.357.556.2]DISCUSSION
There are numerous variant karyotypes seen in TS other than the classic monosomy X. It is important not to confuse iso (X)
chromosome that is 46,X,i(Xq) syndrome with the 45, X classical Turner's syndrome. There are profound cytogenetic
and clinical differences between the two syndromes, which must be borne in mind in the differential diagnosis of amenorrhea and of infertility. (Santana et al., 1977) This isochromosome X consists of the two long arms of the X-chromosome but no short distal arm. Lyon (1961) hypothesized that early in the development of a normal female embryo, random inactivation of one of the two X-chromosomes in each cell occurs which allows the female to have the same amount
[image:3.595.171.425.80.193.2]of X-chromosome material as the average male has. There are certain genes that escape this X inactivation like homeobox gene (SHOX), XIST gene etc. (Santana et al., 1977) These are located predominately in the small regions of homology and pairing that persist on the sex chromosomes called the pseudoautosomal regions (PAR) present on the short distal arm. (Rao et al., 1997) In this way, the normal female has functioning genes from one complete X-chromosome plus functioning genes from the still active short distal arm of the mostly inactivated X-chromosome The short stature phenotype is a result of haploinsufficiency of SHOX (short stature homeobox-containing gene located at Xp22.33) which encodes a transcription factor implicated in skeletal development. Thus, Patients with monosomy X or deletion in Xp or
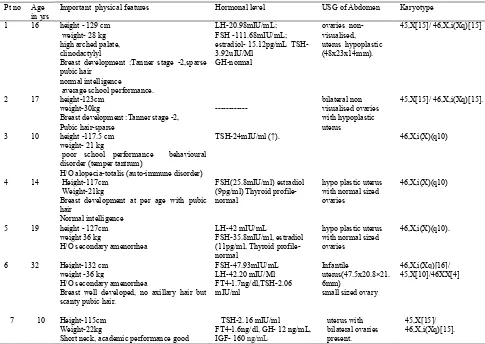
Table 1. Karyotype of study group of patients
No of patients % of patients
Normal 46XX 29 58
45 X (Classical Turner
Syndrome) 11 22
45 X /45 XX( Mosaic Turner
Syndrome) 3 6
46 XXi(q10) 3 6
45 X/46 XXi(q10) 3 6
[image:3.595.54.539.221.565.2]45 X/46 XXi(q10)/46 XX 1 2 50
Table 2. Profile of patients having Isochromosome of long arm of X chromosome
Pt no Age in yrs
Important physical features Hormonal level USG of Abdomen Karyotype
1 16 height - 129 cm weight- 28 kg high arched palate, clinodactylyl
Breast development :Tanner stage -2,sparse pubic hair
normal intelligence average school performance.
LH-20.98mIU/mL; FSH -111.68mIU/mL; estradiol- 15.12pg/mL TSH-3.92uIU/Ml
GH-normal
ovaries non-visualised, uterus hypoplastic (48x23x14mm).
45,X[15]/ 46,X,i(Xq)[15]
2 17 height-123cm weight-30kg
Breast development :Tanner stage -2, Pubic hair-sparse
---
bilateral non visualised ovaries with hypoplastic uterus
45,X[15]/ 46,X,i(Xq)[15].
3 10 height -117.5 cm weight- 21 kg
poor school performance behavioural disorder (temper tantrum)
H/O alopecia-totalis (auto-immune disorder)
TSH-24mIU/ml (↑). 46,X,i(X)(q10)
4 14 Height-117cm Weight-21kg
Breast development at per age with pubic hair
Normal intelligence
FSH(25.8mIU/ml) estradiol (9pg/ml) Thyroid profile-normal
hypo plastic uterus with normal sized ovaries
46,X,i(X)(q10)
5 19 height - 127cm weight 36 kg
H/O secondary amenorrhea
LH-42 mIU/mL
FSH-35.8mIU/ml, estradiol (11pg/ml, Thyroid profile-normal
hypo plastic uterus with normal sized ovaries
46,X,i(X)(q10).
6 32 Height-132 cm weight -36 kg
H/O secondary amenorrhea
Breast well developed, no axillary hair but scanty pubic hair.
FSH-47.93mIU/mL LH-42.20 mIU/Ml FT4-1.7ng/dl,TSH-2.06 mIU/ml
Infantile
uterus(47.5x20.8×21. 6mm)
small sized ovary
46,X,i(Xq)[16]/ 45,X[10]/46XX[4]
7 10 Height-115cm Weight-22kg
Short neck, academic performance good
TSH-2.16 mIU/ml FT4-1.6ng/dl, GH- 12 ng/mL, IGF- 160 ng/mL
uterus with bilateral ovaries present.
45,X[15]/ 46,X,i(Xq)[15].
Normal hormone levels: FSH- 3.50–12.50 mIU/mL, LH-2.40–12.60 mIU/Ml, oestradiol-24.50–195.00 pg/mL,Free thyroxin(FT4)- 0.7-1.9 ng/dl,TSH-0.27–4.20 uIU/Ml, Insulin
isochromosome of one of the long arm of X chromosome(i(Xq)) causes skeletal abnormality like short stature.
Some reports (Sönmez et al., 1997; García et al., 1991; Zinman et al., 1984)have indicated that patients with the 46,X,i(Xq) karyotype have characteristics similar to those observed in classical TS but in a milder form. Those reports claim that the risks for hypothyroidism and mild mental retardation are higher in these patients than in the healthy population. Comparing the patient with isochromosome Xq with individuals who have the 45,X type of TS, the probability of partially developed nipples and mental retardation was higher but the probability of a low-posterior hairline, neck webbing, and hypoplastic nails was lower. However, patients with a deletion of Xp have short stature and congenital malformations. Those with deletion of Xq often only have gonadal dysfunction. (Sönmez et al., 1997) Sybert and McCauley (2004) have reported the 46,X,i(Xq) karyotype in 7% of patients with TS. In our study we have found pure isochromosome Xq in 3 patients and in 3 cases it was mosaic isochromosome Xq. Patient 1 with mosaicism showed few phenotypical features of TS like clinodactylyl and high arched palate, but others isochromosome-Xq didn’t show any morphological stigmata of TS except short stature. Most of the patient with TS show normal intelligence though 70% patient have learning disabilities affecting non verbal perceptual motor and visuo-spatial skills (Ross et al., 2000). The patients with r(X) are at a higher risk of mental retardation, learning difficulties, autistic spectrum disorders, and structural brain abnormalities due to loss of XIST region. (Skuse et al., 2006) We observed no mental retardation in either of these patients but patient no.3 showed very poor school performances and temper tantrum disorders for which she needed psychological counselling by professionals.
TS may be associated with congenital cardiac anamolies (prevalence 17-45%) like coarctation of aorta, bicuspid aortic valve etc or renal malformations with no clear genotype-phenotype correlations. But in our patients we did not find any such congenital malformations. Hypothyroidism is common in 15-30% of patients with TS. There is reported cases of more incidence of auto immune thyroiditis in isochromosome X (García et al., 1991; Chiovato et al., 1996; Medeiros et al., 2000). In our study patient 3 presented with hypothyroidism. She had a history of alopecia-totalis, an auto immune disorder, thus she was advised to have an thyroid scan to rule out auto immune thyroiditis but didn’t turn up with the report. Normal gonadal development needs the ZFX gene in the X chromosome short arm. Although oocyte development requires only a single X chromosome, oocyte maintenance requires two X chromosome. In the absence of a second X chromosome, therefore, oocytes in fetuses and neonates with TS degenerate and their overies atrophy in to streaks of fibrous tissue. Women with an X monosomy, an X long arm isochromosome and short arm deletions commonly present with gonadal dysgenesis due to haploinsufficiency of this gene. (Ogata et al., 2001) Furthermore, some regions in the X chromosome long arm are related with ovarian failure. The presence of an X long arm isochromosome, even in mosaicism with other cell lines, did not present spontaneous menarche. Gonadotrophins may remain suppressed during childhood inspite of ovarian
dysfunction but majority of the patient with i(Xq)have high plasma gonadotrophin (FSH,LH) levels with low estradiol and progesterone level. This hypertropic-hypogonadism phenomenon can be explained with absence of feedback mechanism due to lack of ovarian functions (Zinn and Ross, 2001). In patient 1 and 3 showing such hypertropic-hypogonadism and presented with primary amenorrhea. Patient 3 &7 due to their age not evaluated for these hormonal assay. Thus in a short-stature female with amenorrhea, when FSH is clearly high and the clinical signs of puberty are absent, a pubertal induction with oestrogen should be started. The purpose of this induction is to achieve a similar physical and psychological development as the one occurring in a spontaneous puberty and to establish adequate peak bone mass. However, as oestrogen speed up bone epiphyseal fusion, concurrent oestrogen therapy with growth hormone decreases the effectiveness of GH. So the moment of starting hormone therapy must be coordinated, aiming to achieve maximum growth potential, without unduly delaying the beginning of puberty. Patient 5 & 6 showed secondary amenorrhea, but menstruation always done earlier with hormonal induction.
In addition, an association between inflammatory bowel disease especially ulcerative colitis and i(Xq) genotype was demonstrated. (Manzione et al., 1988) Correlation between acute monocytic leukemia and i(Xq) has also been reported.
(Otokida et al., 1990)
In our study we have found only growth retardation as constant feature with primary amenorrhoea in case 1 &2. We found that the isochromosome i(Xq) form of TS was generally milder than classic TS. A female with short stature, but without typical clinical findings of TS, should be evaluated for this chromosomal form, because 45, X karyotype can be diagnosed at birth due to typical dysmorphic features or cardiac abnormalities, but in isochromosome-X diagnosis may be delayed until childhood, adolescence or unfortunately until adulthood while evaluation for short stature ,pubertal delay, primary amenorrhea and infertility. Early diagnosis is an important aspect of ideal treatment for these variant type of TS patients because growth hormone supplementation in proper time can help in achieving normal height at per age followed by sex hormone supplementation for overcoming ovarian dysfunction.
Acknowledgement
We acknowledge constant cooperation and help of Swami Satyadevananda, (secretary of VIMS, Kolkata) during the study period.
REFERENCES
Donaldson MD, Gault EJ, Tan KW, Dunger DB. Optimising management in Turner syndrome: from infancy to adult transfer Arch Dis Child. 2006 Jun;91(6):513-20.
Jacobs P, Dalton P, James R, et al. Turner syndrome: A cytogenetic and molecular study. Ann Hum Genet., 1997; 61:471–483
Graham GE, Allanson JE, Gerritsen JA. Sex chromosome abnormalities. Rimoin DL, Connor JM, Pyeritz RE, Korf BR (eds). Principles and Practice of Medical Genetics. 5th ed. Edinburgh, Scotland: Churchill Livingstone; 2007:1038–1057.
Sybert VP, McCauley E. Turner’s syndrome. N Engl J Med., 2004;351:1227–1238.
Morgan T. Turner syndrome: Diagnosis and management. Am Fam Physician., 2007;76:405–410.
Lanes R.Long-Term Outcome of Growth Hormone Therapy in Children and Adolescents. Treat Endocrinol., 2004; 3(1) : 53-66.
Santana JAM, Gardner LI, Neu RL. The isochromosome-X syndrome [46,Xi(Xq)]: Report of three cases with review of the phenotype. ClinPediatr (Phila). 1977;16:1021–1026. Brown, C. J. and Willard, H. F. Localization of a Gene that
Escapes....Short Arm: Implications for X Inactivation. American Journal of Human Genetics,1990; 46: 273-279. Rao E, Weiss B, Fukami M, Rump A, Niesler B, Mertz A et
al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet., 1997 May;16(1):54-63.
Sönmez S, Sönmez Y, Öztas S, et al. IsochromosomeXq in a girl having delayed puberty. Journal of TurgutÖzal Medical Center, 1997;4:109–111.
García CB, Robles CP, González VA, et al. Hypothyroidism and isochromosome X in Turner’s syndrome [in Spanish]. An EspPediatr., 1991;34:161–162.
Zinman B, Kabiawu SI, Moross T, etal. Endocrine, cytogenetic and psychometric features of patients with X-isochromosome 46, X, i(Xq) Turner’s syndrome: A preliminary study in nine patients. Clin Invest Med., 1984;7:135–141.
Ross JL, Zin A, McCauleyE.Neurodevelopmental and psychosocial aspects of Turner Syndrome. Ment Retard DevDisabil Res Rev., 2000; 6:135-41.
Skuse D, Elgar K,Morris E,Turner C. Ring-X Chromosomes:Their cognitive and behavioural phenotype. Ann Hum Genet., 2006; 64: 295-305.
García CB, Robles CP, González VA, et al. Hypothyroidism and isochromosome X in Turner’s syndrome [in Spanish]. An EspPediatr., 1991;34:161–162.
Chiovato L, Larizza D, Bendinelli G, Tonacchera M, Marinó M, Mammoli C, Lorini R, Severi F, Pinchera A: Autoimmune hypothyroidism andhyperthyroidism in patients with Turner's syndrome. Eur J Endocrinol., 1996, 134:568–575.
Medeiros CC, Marini SH, Baptista MT, Guerra G Jr, Maciel-Guerra AT: Turner’s syndrome and thyroid disease: a transverse study of pediatric patients in Brazil. J Pediatr Endocrinol Metab., 2000, 13:357–362.
Ogata T, Muroya M, Matsuo N, et al. Turner syndrome and Xp
deletions: clinical and molecular studies in 47 patients. J
Clin Endocrinol Metab., 2001;86:5498-508.
Zinn AR, Ross JL. Molecular analysis of genes on Xp controlling Turner syndrome and premature ovarian failure
(POF). Semin Reprod Med., 2001;19:141-6.
Manzione NC, Kram M, Kram E, Das KM. Turner's syndrome and inflammatory bowels disease: a case reportwith
immunologic studies. Am J Gastroenterol., 1988;83(11):
1294-7.
Otokida K, Oshira K, Ishikawa M, et al. A case of Turner
syndrome with the karyotype of 45, X/46, X, i(Xq)associated with the acute monocytic leukaemia.
Tohoku J Exp Med., 1990; 161(1): 19-24