R E S E A R C H
Open Access
Pattern matching through Chaos Game
Representation: bridging numerical and discrete
data structures for biological sequence analysis
Susana Vinga
1,2*, Alexandra M Carvalho
3,4, Alexandre P Francisco
1,4, Luís MS Russo
1,4and Jonas S Almeida
5Abstract
Background:Chaos Game Representation (CGR) is an iterated function that bijectively maps discrete sequences into a continuous domain. As a result, discrete sequences can be object of statistical and topological analyses otherwise reserved to numerical systems. Characteristically, CGR coordinates of substrings sharing anL-long suffix will be located within 2-Ldistance of each other. In the two decades since its original proposal, CGR has been generalized beyond its original focus on genomic sequences and has been successfully applied to a wide range of problems in bioinformatics. This report explores the possibility that it can be further extended to approach
algorithms that rely on discrete, graph-based representations.
Results:The exploratory analysis described here consisted of selecting foundational string problems and refactoring them using CGR-based algorithms. We found that CGR can take the role of suffix trees and emulate sophisticated string algorithms, efficiently solving exact and approximate string matching problems such as finding all palindromes and tandem repeats, and matching with mismatches. The common feature of these problems is that they use longest common extension (LCE) queries as subtasks of their procedures, which we show to have a constant time solution with CGR. Additionally, we show that CGR can be used as a rolling hash function within the Rabin-Karp algorithm.
Conclusions:The analysis of biological sequences relies on algorithmic foundations facing mounting challenges, both logistic (performance) and analytical (lack of unifying mathematical framework). CGR is found to provide the latter and to promise the former: graph-based data structures for sequence analysis operations are entailed by numerical-based data structures produced by CGR maps, providing a unifying analytical framework for a diversity of pattern matching problems.
Background
Biological sequence analysis remains a central problem in bioinformatics, boosted in recent years by the emer-gence of new generation sequencing high-throughput techniques [1,2]. With the advances of new technologies, the need for efficient algorithms for string representa-tion and processing is increasing continuously.
Sequence representation plays a particular role in this regard, being the first step that both enables and condi-tions subsequent data processing, and has been the object of several analytical approaches over the last years [3]. The
development of string processing algorithms based on different data structures, particularly suffix trees and suffix arrays, has been increasing steadily [1,4]. The classical ver-sion of these structures enhances the functionality of the underlying sequence by efficiently supporting several look-up procedures [4]. In particular, these structures can be used to locate substrings within the index sequence, which in turn yields efficient algorithms for complex problems such as the longest common substring, initially thought to have no linear time solution. Unfortunately, the speed up obtained from these structures comes with an heavy mem-ory cost. This cost has been extensively researched, the most successful trend consisting of using data compres-sion techniques, such as Lempel-Ziv data comprescompres-sion, Burrows-Wheeler transform, and local compression * Correspondence: [email protected]
1
Instituto de Engenharia de Sistemas e Computadores: Investigação e Desenvolvimento (INESC-ID), R. Alves Redol 9, 1000-029 Lisboa, Portugal Full list of author information is available at the end of the article
methods, achieving dramatic improvements in efficiency (see [5] for a survey). Moreover, these theoretical results allowed for the development of compressed aligners that have significantly reduced the resources necessary for sequence alignment [6-8], namely for aligning reads from pyro-sequencing platforms [9].
In parallel to these efforts, a particular prolific techni-que at engendering novel setechni-quence analysis algorithms is theChaos Game Representation(CGR), based onIterated Function Systems(IFS), firstly proposed more than two decades ago [10]. This compact, lossless and computa-tionally efficient representation allows the visualization of biological sequences and patterns, with a convenient visual appearance. In fact, CGR captures the patterns arising from distinct L-tuple compositions in DNA sequences [11], enabling whole genome representation and visualization [12]. It was also shown that CGR repre-sents a generalization ofgenomic signatures [13,14], allowing to characterize different species by associating them with distinctive sequence statistics [12].
In addition, CGR can be interpreted as a generalization of Markov Chain models [15] and provides tools for the alignment-free analysis and comparison of biosequences [16], namely for DNA entropy estimation [17,18]. Some other applications of CGR include the problem of HIV-1 subtype classification [19], detection of persistent dynamic of human spike trains [20], horizontal transfer detection [21], phylogeny and extraction of representative motifs in regulation sequences [22], to name a few.
Although CGR has been successfully applied to a wide range of problems, its strength for general string matching problems is, so far, mostly unexplored. In this work we propose CGR as a solution for exact and approximate string matching problems, showing that this representa-tion might bring useful algorithmic tools for biological sequence analysis and more general applications.
Methods
We start this section by introducing some notation and recalling the definition and relevant properties concern-ing CGR. Thereafter, we embed CGR maps in the Can-tor set in order to elicit longest common suffixes in an efficient and unambiguous way. As we shall see in the Results section this enables the retrieval of longest com-mon extension queries in constant time, which in turn yield efficient algorithms for more complex string matching problems.
LetSdenote a string of sizeN. In all what follows,S[i] represents thei’th symbol ofS, with 1≤i≤N. EachS[i] is a DNA symbol representing each nucleotide or base (A, C, G, T), i.e.,S[i]ÎΣ= {A, C, G, T}. Moreover,S[i .. j] is thesubstringofSthat starts at thei’th and ends at the
j’th positions ofS, with 1≤i≤j≤N. Thelengthof sub-stringS[i..j] is given by|S[i..j]|=j - i + 1. Furthermore,
S[i..] denotes thesuffixofSthat starts at thei’th position, i.e.,S[i..] =S[i..N]. Similarly,S[..i] denotes theprefixofS
that ends at thei’th position, i.e.,S[..i] =S[1..i].
Chaos Game Representation (CGR) definition
The CGR iterative algorithm is constructed on a square in IR2 where each of its vertex is assigned to a DNA symbol or base (A, C, G, T). For a given DNA sequence
Sof length N, CGR maps each prefix S[..i] onto a point
xiÎIR2following the iterative procedure:
x0=
1 2,
1 2
xi=xi−1+1
2(yi−xi−1),i= 1,. . .,N whereyi= ⎧ ⎪ ⎪ ⎨ ⎪ ⎪ ⎩
(0, 0) ifS[i] =A
(0, 1) ifS[i] =C
(1, 0) ifS[i] =G
(1, 1) ifS[i] =T. (1)
The initial point x0 in the original formulation [10] was taken as the square center, i.e., x0=12,12. Alterna-tively, this point could be chosen randomly on the square, x0 ~Unif([0, 1]2), or calculated as the last sym-bol x0=xNby differently seeding the iterative function, providing symmetry adequate for circular genomes [23]. In addition, thecontraction ratioof the original formu-lation is equal to r= 1
2, which means that each iteration
transforms the whole square into a sub-quadrant with length half the size. More generally, this value can be any number from 1
2 ≤r<1 to preserve bijectivity.
An alternative and non-recursive expression forxias a function of all symbolsy1,...,yi, contraction ratio r, and initial pointx0, is given by
xi= (1−r)ix0+r
i
k=1
(1−r)k−1yi−k+1
= (1−r)ix0+r
i
k=1
(1−r)i−kyk, i= 1,. . .,N. (2)
The generalization of CGR to higher-order alphabets was defined, in a more natural way, by extending the original CGR square to an hypercube [24], which can be bijectively mapped to 2D polygons by increasing the value of r [25]. The dimensiondof the hypercube will depend on the length of the alphabet |Σ| asd= log2 |Σ| and all the remaining properties are maintained.
CGR properties
One important property of CGR is the convergence of sequence coordinates in the space [0,1]2when the pre-fixes they represent share a common suffix. In fact, CGR maps prefixes with shared suffixes close to each other reflecting the property that, wherever the sequence context may be, the same suffix will be always mapped in the same region of CGR.
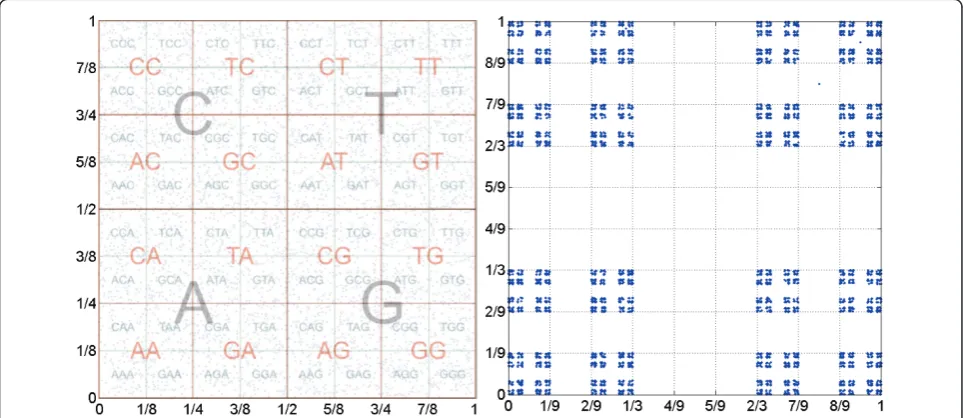
of 2 in each common symbol in their suffixes. One con-sequence of this result is that the distance between the coordinates being less that 2-L constitutes a necessary condition for sharing a L-long suffix. Another foregone conclusion is that CGR maps can be divided and labelled according to the corresponding substring, i.e., each substring is mapped onto a sub-square, creating a fractal-like structure. This allows the retrieval of Markov Chain transition probability tables [15], as exemplified graphically in Figure 1a. This result is not sensitive to the initial condition or pointx0 chosen, since the Lyapu-nov exponent is negative.
More generally, in order to extract transition probability estimates for anL-order Markov chain, the interval [0,1] should be divided in 2L+1subintervals. The number of points in each sub-square thus created is then counted, which is the same as extracting the number of (L +1)-nucleotides. Extensions of this property allowed the esti-mation of Variable Length Markov Models (VLMM) [26,27] and time series forecasting byFractal Prediction Machines(FPM) [28]. For other ratiosrand initial points
x0these properties are all maintained. More recently, CGR further enabled a novel type of sequence analysis that associates its fractal geometry properties with biological structures [29].
Constant-time longest common suffix via Cantor set embedding
As discussed previously CGR coordinates of prefixes sharing anL-long suffix are located within 2-Ldistance of
each other. However, there are prefixes not sharing an
L-long suffix that are also located within the same dis-tance. We conclude that being in that neighborhood is not a sufficient condition to determine a commonL-long suffix, as in some borderline cases the common suffix crudely taken from this fact is longer than the real one. In this section we address this shortcoming aiming at finding an efficient procedure to elicit longest common suffixes within CGR maps.
Intuitively, having not noticed that the aforementioned condition is only necessary, we would believe that the size of the longest common suffix ofS1[..i] andS2[..j] is given by the greatestℓ for which theL∞-norm
|xS1
i −x
S2
j |∞= max b=1,2{|x
s1
i:b−x S2
j:b|}
is still less than 2-ℓ, where xi = (xi:1, xi:2) for all i. Observe that the L∞-norm corresponds simply to the maximum distance on the two CGR dimensions. Find-ing suchℓcan be accomplished in constant time by the mean of a logarithm operation given by
−log2(|x
S1
i −x
S2
j |∞)
−1. (3)
To see why Eq. (3) fails to compute the longest com-mon suffix, letS1 =AT and S2 =TA, therefore, N= 2. Moreover,i=j= 2. Clearly, the longest common suffix betweenS1and S2 is 0, as they share no common suffix. However, using Eq. (3) we obtain
Figure 1CGR representation. Chaos Game Representation (CGR) map of random DNA sequences of lengthN= 5,000. The first panel
represents the original CGR proposal with contraction parameter p= 12and the superimposed sub-quadrants of the suffix labels. The second
p= 2 3
This indicates an incorrect common suffix of length ℓ = 1 between S1 and S2. If these strings are further extended toS1=A(A)Tand S2 =T(T)A, where (a) is a sequence of symbol aÎ Σ as long as wanted, then the corresponding CGR coordinates xS1
N and x
S2
N will further approximate each other in the map and Eq. (3) will result inℓ=N- 1, whereas their longest common suffix remains 0. We refer the intended reader to Additional file 1 where details on this analysis can be found.
The core of the problem lies on Eq. (3). Start by noticing that the logarithm in Eq. (3) corresponds to the number of leading zeros in the fractional part of |xS1
i −x
S2
j |∞when
thisL∞-norm is seen in binary numeral system (base 2).
Intuitively, these leading zeros, sayL, represent a match betweenS1[i-L+ 1...i] andS2[j - L+ 1...j]. The shortcom-ing of Eq. (3) is that theL∞-norm introduces an unex-pected problem with the carry mechanism. To better see why, assume again thatS1=ATandS2=TA, soN= 2. In this case we can write the CGR coordinates xS1
2 and x
S2 2 in
base 2 as xS1
2 =(0.101, 0.101) and x
S2
2 =(0.011, 0.011)
When computing the difference xS1 2 −x
S2
2 between the
coordinates we get
0. 1 0 1
− 0. 01 1 1
0. 0 1 0
,
and we come across a carry that masks the real dis-similarity between the last characters of stringS1 andS2 (highlighted in bold). Indeed, the difference xS1
2 −x
S2 2
indicates that the first character of S1 and S2 match, which is not the case.
In order to solve the longest common suffix in con-stant time we must be able to compute the difference between CGR coordinates without incurring the penalty of the carry mechanism. We achieve this by embedding the CGR coordinates in theCantor set [30,31]. For that, we use base 3, instead of base 2, admitting a numeral representation consisting entirely of 0’s and 2’s (never using the digit 1). A way to achieve this is to consider that usual CGR coordinates are expressed in base 3 by replacing all 1’s with 2’s. Such CGR coordinates can be computed similarly as in Eq. (2) by
xi= 2 × 3−i
1 3,
1 3
+
i
k=1
2×3−kyi−k+1
= 2 × 3−i
1 3,
1 3
+
i
k=1
2 × 3k−i−1yk, i= 0,. . .,N.
(4)
In this case, the difference xS1 2 −x
S2
2 in the previous
example becomes
0. 2 0 2
− 0. 01 2 2
0. 1 1 0
indicating, as expected, that strings S1 and S2 mis-match in the first symbol.
This transformation generally leads to the desired scalability because the carry-on mechanism does not change the difference between the operands in the first bit where the difference takes place. This is a mandatory practice for the base-3 logarithm log3(|xS1
2 −x
S2
2 |∞)
cor-rectly identifying the first bit where the difference between the coordinates is found. In the following, we show this fact in detail. Start by noticing that when dealing with the L∞-norm of the difference we only have to cope with differences where the left operand is greater than the right one. In that case, the dissimilarity may take place exactly in the first digit of the difference. If so, a carry-on may, or may not, take place just before moving to the second digit of the difference. Either way we end up with the same base-3 logarithm, as in both cases the first digit of the difference is greater than 0 (highlighted in bold), i.e.,
0. 2 . . . 2
− 0. 0 . . . 2 0. 2 . . . 0
and
0. 2 . . . 2
− 0. 01 . . . 2 0. 1 . . . 0 .
On the other hand, if the dissimilarity does not take place in the first digit of the difference, we conclude that the first bit of the difference greater than 0 will be exactly the first bit where the mismatch occurs, either with a carry-on or not. Indeed, assuming that the firstk
bits of the difference xS1 2 −x
S2
2 are similar we have that
0.b1 . . . bk 2 . . . 2
− 0.b1 . . . bk 0 . . . 2
0. 0 · · · 0 2 . . . 0
and
0.b1 . . . bk 2 . . . 2
− 0.b1 . . . bk 01 . . . 2
0. 0 · · · 0 1 . . . 0
,
correctly concluding that a mismatch occurs at the (k+ 1)’th bit of the difference and, therefore,S1andS2have a longest common suffix of sizek.
In addition, we show that using CGR coordinates embedded in the Cantor set as in Eq. (4) we achieve a sufficient and necessary condition to compute longest common suffixes in constant time. For the sake of sim-plicity, and without loss of generality, we assumeN=M
andi=j.
Theorem.Let S1 and S2 denote two strings of size N.
We have that xS1
i −x
S2
i ∞<3−Lif and only if S1[i - L+
Proof:We start by showing that if xS1
i −x
S2
i ∞<3−L
thenS1[..i] andS2[..i] have a commonL-length suffix. This part of the proof follows by contradiction. Thus,
assume that xS1
i −x
S2
i ∞<3−L and thatS1[i - L+ 1..i]≠
S2[i - L+ 1..i]. IfS1[i - L+ 1..i]≠S2[i - L+ 1..i] then, in base 3 numeral representation,
we have that xSd
i:c= 0.b1. . .bk2. . . and
xS3−d
i:c = 0.b1. . .bk0. . ., for some 1≤k < Landc, d= 1, 2. Then,
3−(k+1)<xSd i:c−x
S3−d
i:c <2 × 3−
(k+1)<
3−k.
This contradicts the hypothesis that
xS1
i −x
S2
i ∞<3−L, concluding this part of the proof.
We now proceed to prove that ifS1[..i] andS2[..i] have
a common L-length suffix then xS1
i −x
S2
i ∞<3−L.
Since by hypothesis S1 andS2 have a commonL-length suffix we know that yS1
k =y
S2
k for all i-L+1 ≤ k ≤ i. Hence, by Eq. (4) we have that
xS1
i −x
S2
i = 2×
i−L
k=1
3k−i−1(yS1
k −y
S2
k ).
TheL∞-norm of this difference is then given by
|xS1 i −x
S2 i|∞=
2×
i−L
k=1
3k−i−1(yS1 k −y
S2 k)
∞
≤2×(3−L−1+ 3−L−2+· · ·+ 3−i)<3−L.
This concludes the proof.
Interestingly, this representation corresponds to a var-iant of CGR that uses a contraction ratio ofr= 23, instead
of the originalr= 12, and an initial point in the set
attrac-tor x0= 23. In fact, this result is achieved by using a simi-lar recursive expression as the one given by Eq. (1):
x0=2 3,23
xi=xi−1+2
3(yi−xi−1),i= 1,. . .,N whereyi=
⎧ ⎪ ⎪ ⎨ ⎪ ⎪ ⎩
(0, 0) ifS[i] =A
(0, 1) ifS[i] =C
(1, 0) ifS[i] =G
(1, 1) ifS[i] =T. (5)
The graphical comparison between these two versions is given in Figure 1. The sub-quadrants in the first case are contiguous and, therefore, the closeness in this representation is a necessary but not a sufficient condi-tion for sharing the same suffix. On the contrary, in the second representation, the sub-quadrants are separated by at least 3-Land so the condition becomes sufficient, as the border effect is absent for everyL-long suffix.
Results
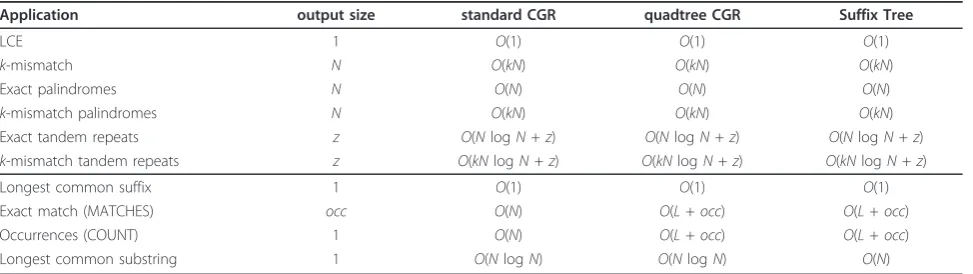
In this section we present several exact and approximate string matching problems, along with their CGR solu-tion. These include the longest common extension pro-blem, as well as finding all palindrome and tandem repeats, exact and approximate string matching, and longest common substring. In addition, we show how CGR can be used as a rolling hash for the Rabin-Karp string search algorithm. A study of the time complexity attained by these algorithms is given throughout this section and a summary in provided in Table 1.
All the code for both Mathematica® and JavaScript implementations is made available at http://cgrsuffix. github.com/.
CGR data structures and implementation
CGR was implemented using two different data struc-tures, a standard floating-point matrix and a quadtree-like structure. In this section we describe these imple-mentations along with the required notation.
Standard CGR index
The standard implementation uses a floating-point representation that calculates all the coordinates itera-tively using Eq. (5). In this implementation, a stringSof size Nis represented in memory by a floating-point matrix of sizeN ×2. This corresponds to aN-size array
Table 1 Time complexity of different applications
Application output size standard CGR quadtree CGR Suffix Tree
LCE 1 O(1) O(1) O(1)
k-mismatch N O(kN) O(kN) O(kN)
Exact palindromes N O(N) O(N) O(N)
k-mismatch palindromes N O(kN) O(kN) O(kN)
Exact tandem repeats z O(NlogN+z) O(NlogN+z) O(NlogN+z)
k-mismatch tandem repeats z O(kNlogN+z) O(kNlogN+z) O(kNlogN+z)
Longest common suffix 1 O(1) O(1) O(1)
Exact match (MATCHES) occ O(N) O(L+occ) O(L+occ)
Occurrences (COUNT) 1 O(N) O(L+occ) O(L+occ)
Longest common substring 1 O(NlogN) O(NlogN) O(N)
of pairs, denoted by xS= (xS
i, . . . ,xSN), where the i’th entry xSi = (xSi:1,xSi:2) is accessed inO(1) time, for all 1≤
i≤N.
This data structure is built in optimal time, that is, in time linear in the length of the stringS. Indeed, it takes
O(N) time to build the matrix and each entry of the matrix is computed inO(1) time. This constant time is achieved as, for all 1 ≤ i ≤ N, coordinate xSi can be computed from coordinate xSi−1 as in Eq. (5) by a fixed number of sum operations.
Depending on the application at hand we may need to build one or two standard CGR maps. These CGR maps may correspond to the direct DNA string as well as to its reverse. In addition, the complement or reverse com-plement may also be used. The reverse string of S is denoted by Sr and the reverse complement of S is denoted bySrc. If the string Srcis reversed once again, the complement string Sc is obtained. Finally, given a string S, we call the CGR mapxS thedirect CGR index, the CGR map xSr
thereverse CGR index, the CGR map
xSrc
the reverse-complement CGR index, and the CGR map xSc
thecomplement CGR index. Quadtree-based CGR index
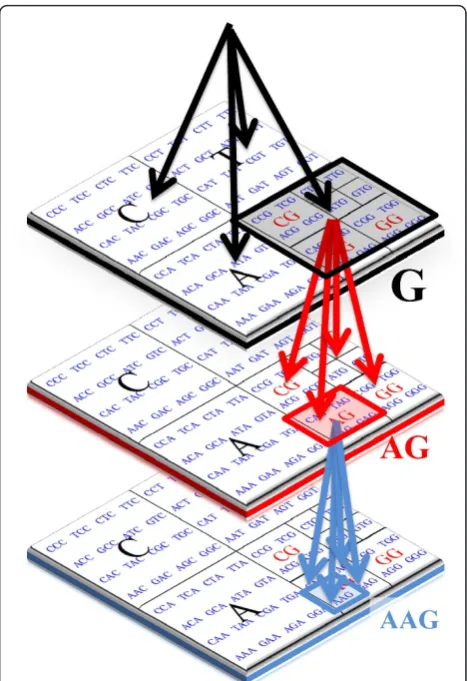
Since in its simplest form a CGR map is just a set of coordinates on a two-dimensional plan, we can use a quadtree to index such points [32]. For this index, a tree data structure is used where each node has at most four children. Each child node represents a sub-quadrant of its parent node containing an extension of the parent prefix. Assuming Σ = {A, C, G, T}, we identify each child byA, C, Gand T. Given a stringSwith size N, at mostNnodes are stored in the quadtree as follows: for each prefixS[1..i] of S, we start inserting its reverse on the quadtree until we reach a leaf, where we insert a new node and stop. For instance, let S =GACGA and let us start with an empty quadtree [,,, ]. After inserting the prefixG, the quadtree becomes [,, 1: [,,,],] where the symbolS[1] is the unique symbol in quadrantG. After inserting prefixes GAandGAC, the quadtree becomes [2: [,,,], 3: [,,,], 1: [,,,],] as S[1], S[2] and S[3] are the unique points in quadrants G, A and C, respectively. But, after inserting the prefix GACG, the quadtree becomes [2: [,,,], 3: [,,,], 1: [, 4: [,,,],,],] as the quadrantG
has a second pointS[4] in the subquadrant GC. Simi-larly, after inserting the prefix GACGA, the quadtree becomes [2: [,, 5: [,,,],], 3: [,,,], 1: [, 4: [,,,],,],], where the quadrantAhas a new second point in subquadrantAG.
Note that quadtrees as described are justcompressed triesofSprefixes, also known asCGR-trees, described as quaternary trees related todigital search treesproposed by Cenac et al. [33]. Moreover, the use of a quadtree to index the CGR coordinates is equivalent to a suffix tree,
up to an inversion of the text (because of the prefix/suffix duality). Hence, although as general search trees, trees may degenerate and its construction becomes quad-ratic on string size, we can improve such construction to linear time in worst case using Ukkonen’s algorithm [34] after an inversion of the text, as expected. For larger alphabets higher order trees, such asoctrees, can be considered.
Web-based JavaScript implementation
Along with the Mathematica®implementation, a Java-Script version is also provided, where an efficient binary representation of CGR coordinates is used, thus eliminat-ing floateliminat-ing point operations. Additionally, this implemen-tation also uses quadtrees for indexing CGR maps, improving search complexity by at least one order of mag-nitude when compared with traditional implementations.
We choose JavaScript as underlying language so that our implementation can be used in the Web distributed com-putational environment. The JavaScript interpreters, such as those found on modern browsers, include high perfor-mance features such as runtime compilation, achieving performances usually associated with scientific computing environments. Furthermore, it natively supports code migration (through the SCRIPT element of the browsers Document Object Model) which provides a seamless mechanism for code distribution and reuse.
Longest common extension
Given two stringsS1andS2, each one with lengthNand
M, and a long sequence of position pairs, the longest common extension(LCE) problem consists in finding, for a given query pair (i, j), the length of the longest prefix ofS1[i..] that matches a prefix ofS2[j..].
This problem is directly related with the longest com-mon suffix described in the previous section, up to an inversion of the strings. Therefore, to address LCE queries with standard CGR indexes it suffices to search for shared suffixes in the reverse CRG indexes ofS1andS2, as that suffixes would represent prefixes in the direct indexes. The position pair (i, j) needs to be updated accordingly, to (N-i+ 1, M- j+ 1), in order to reference the correct positions in the reverse strings. With suffix/prefix duality problem solved we resort on CGR maps embedded in the Cantor set to perform LCE queries.
The pseudo-code to find the LCE ofS1[i..] and S2[j..], with|S1|=Nand|S2|=M, is given in Algorithm 1.
Algorithm 1 Longest common extension (LCE) ofS1
andS2for position pair (i, j)
LCE(standard CGR index entry xS
r
1
N−i+1, standard
CGR index entry xSr2
M−j+1)
1. return −log3(|xSr1
N−i+1−x Sr
2
M−j+1|∞)
This single step algorithm makes each LCE query answered in constant time. Many string algorithms use LCE as a subtask that needs to be addressed efficiently and will therefore benefit from this result, as shown in the following sections.
Common operations on strings
In this section, several common operations on strings are analyzed and a comparison between CGR and tradi-tional data structures is given when appropriate. This analysis focus on how each operation can be achieved through different CGR implementations, along with its respective time complexity.
Given a stringS of size N and a patternP of size L, searching for occurrences ofP withinSis probably the most common operation on strings. This problem, named MATCHES, has been utterly studied and there are well known algorithms to solve it in linear time on the length ofSand P. The problem becomes more chal-lenging if we are given a stringSand we want to search, possible online, for a set of patterns within S. In this case, the preprocessing and indexing ofS can greatly improve the search time as we just process S once, which is not the case if we use algorithms such as Knuth-Morris-Pratt [35] or Aho-Corasick [36]. In this context, the stringS needs to be indexed and standard or quadtree-based CGR indexes can be used.
If a standard CGR index is used, the occurrences of P
in S can be found by scanning S with LCE queries to match P in each suffix S[i..], with 1 ≤ i ≤ N - L + 1. More precisely, if the LCE query ofS andPfor a given position pair (i, 1) returnsLthenP matchesS[i..i +L
-1]. The algorithm for computing all matches of a pat-tern Pin a string Sis given as Algorithm 2. Recall that we need to use the reserve CGR indexes ofS andP to tackle suffix/prefix duality and all CGR coordinates are embedded in the Cantor set, thus, defined as in Eq. (5). Concerning time complexity, Step 2 takesO(N-L) and both Step 3 and Step 4 take O(1), hence, the overall complexity of MATCHES isO(N) asL < < N.
Algorithm 2MATCHES with standard CGR index
MATCHES(standard CGR index xSr
, patternP) 1. let xPr
= (xPr
1,. . .,xP
r
L ) be the CGR coordinates forPr
2. for (ifrom 1 toN-L+ 1) 3. let k = LCE (xSr
N−i+1, xP
r
L)//longest common prefix ofS[i..] andP[1..]
4. if (k=L) thenPoccurs inS[i..i+L -1]
Although standard CGR indexes can be space efficient, as we only need to store a coordinate for each text posi-tion, the MATCHES operation is not as efficient as with
suffix tree indexes. Indeed, with suffix trees this opera-tion takes only O(L +occ) time, whereocc is the num-ber of occurrences of P in S (usually, occ < < N). Notwithstanding, we can improve the running time of MATCHES by using a quadtree-based CGR index, instead of a standard one. For this purpose, consider a quadtree for the CGR map of S. Given a patternP, we take its reversePrand we descend on the quadtree until we either reach the end ofPror a leaf of the quadtree. Along the descendent path we must check for each internal node if their CGR coordinates are equal to those of P, which takes constant time as in Algorithm 2. If this check is true, we must report an occurrence. If we reach the end of Pr and we are at nodenwith chil-dren, we must also check every node in the subtree rooted at n as each one matches the pattern. Hence, searching for a patternPtakes linear time on the size of
P plus the number of its occurrences in S, i.e.,O(L+
occ) as in suffix trees.
Another common operation on strings is COUNTS. It consists on determining the number of occurrences of a given patternPin a stringS. This can be easily achieved by adapting the MATCHES operation, for both standard and quadtree-based CGR indexes, resulting in the same time complexity. The only difference is that instead of outputting the occurrences a counter is used to retrieve the total number of occurrences ofPinS.
Other common operations on strings include LONG-EST COMMON SUFFIX, IS PREFIX and IS SUFFIX. The LONGEST COMMON SUFFIX was addressed in the Cantor set description, achieving constant time complexity as LCE. Given two stringsSandP, with |S|
=N, |P|=NandL < N, the IS SUFFIX operation con-sists in checking if Pappears as a suffix ofS. This can be done by computing the LCE for coordinates xPL and
xSN. The answer to IS SUFFIX is true if and only if the LCE query returns L, otherwise the answer is false. The IS PREFIX operation consists in checking if P appears as prefix of S and it can be easily done by reversing bothSand Pand applying the previous operation. Since both IS SUFFIX and IS PREFIX operations rely solely in LCE, they achieve constant time complexity as long as the needed CGR indexes are already computed.
We conclude this section by discussing four new operations that are specific to CGR but related with relevant operations in suffix trees (namely, SLINK and WEINERLINK). As we shall see later these operations provide a way for performing incremental hashing, an approach used for Rabin-Karp string matching algo-rithm. The LEFT DELETION of a string S of sizeNis the substring ofS from which the first letter was taken, that is, given S =aP, the LEFT DELETION of S is P. Given the last CGR coordinate ofS, denoted by xS
new CGR coordinate corresponding to the LEFT DELE-TION ofSis given by
xPN−1=xSN−r(1−r)N−1(yS1−x0). (6)
Moreover, the LEFT INSERTION compares what hap-pen when we add a symbol before the string, that is, given a stringS the LEFT INSERTION ofainS isQ=
aS. Given the last CGR coordinate ofS, denoted by xS N, the new CGR coordinate corresponding to the LEFT INSERTION ofainSbecomes
xQN+1=xSN+r(1−r)N+1(yQ1 −x0).
Clearly, this operations are computed in constant time. Finally, we recall that symmetric operations of RIGHT INSERTION and RIGHT DELETION can be straightforwardly computed from the iterative definition of CGR in Eq. (1) also withO(1) time complexity.
The examples presented in this section illustrate the algorithmic equivalence between CGR and suffix tress. Graphically, this can be observed through the represen-tation of both data structures, given in Figure 2.
Finding palindromes and tandem repeats
This section presents two applications of CGR based on exact string matching that rely on LCE described above. A nucleotide sequenceS is said to be a(complemented) palindromeif it is equal to its reverse complement. In this case, the first half ofSconverted to its reverse com-plement is equal to the second half of S. If the two halves of the palindrome are not adjacent it is said to be aseparated palindrome. Moreover, the palindrome is said to have aradiuscorrespondent to the size of each half. Palindromic sequences play an important role in molecular biology. They are, for instance, specifically recognized by many restriction endonucleases [37] and usually associated with methylation sites [38].
The problem of finding all palindromes focus on uncovering all maximal complemented palindromes from a nucleotide sequence. By maximal it means that only the bigger palindrome is extracted. For instance, the sequence TTATAA has a palindrome of radius 1 (AT), a palindrome of radius 2 (TATA), and a palin-drome of radius 3 (TTATAA), but only the latter is maximal.
The algorithm to find all palindromes uses, as subtask, forward and backward LCE queries from the midposi-tions of potential palindromes. The backward query cor-responds, actually, to a forward query over the reverse complement ofS. The detailed pseudo-code of this algo-rithm can be found in Addition file 2. Since each of the aforementioned extensions can be performed over a reverse and a complement CGR indexes in constant
time, the algorithm takesO(N) time, corresponding to the time needed to cover all stringS.
In practice, palindromes are often separated over the DNA sequence. If the gap between the two halves of the palindrome is fixed, it happens to be a simple variant of the previous problem. Therein, forward and backward extensions are performed in positions apart of each other (Additional file 2), a procedure that is again solved in linear time. The general case of finding all separated palindromes, without a fixed gap, is more complex as it is equivalent to the problem of finding all inverted repeats. As expected, aninverted repeatis a sequence of nucleotides that is the reverse complement of another sequence that is found further downstream the DNA sequence. These sequences are usually transposon boundaries [39,40].
Another major application that benefits from con-stant-time LCE queries is finding all tandem repeats in a nucleotide sequence. Tandem repeats are adjacent copies of a same DNA subsequence. They usually
consist of short copies of 2-6 base pairs in length repeated throughout all eukaryotic genomes. Tandem repeats often represent important control sequences, such as upstream promoter sequences [41]. In addition, they have been used in forensic science for determining parentage [42].
The problem entailing tandem repeats focus in locating all tandem repeats in a nucleotide sequence. For instance, the sequenceTTATTAhas a tandem repeatTTof radius 1 and a tandem repeatTTATTAof radius 3, both starting in the first position of the nucleotide sequence. It also con-tains a tandem repeatTTof radius 1 at position 4.
The algorithm to find for all tandem repeats was first proposed by Landau and Schmidt [43]. It is a divide and conquer algorithm that recursively break down the problem of finding tandem repeats into sub-problems, until these become simple enough to be solved directly. These simpler problems can be solved through two LCE queries to elicit the presence of a tandem repeat. The time complexity of the algorithm isO(NlogN+z) wherezis the number of tandem repeats inS. The details of the algorithm, which requires the direct and reverse CRG indexes to be built in a pre-processing phase, are presented in Additional file 2.
Approximate string matching
Thek-mismatchproblem is an approximate matching pro-blem where k mismatches are allowed. Approximate matching is a mandatory task in molecular biology as DNA sequence patterns are usually described in an approximate way. This makes way for more relaxed ver-sions of palindrome and other repeat problems. Landau and Vishkin [44] and Myers [45] were the first to present anO(kN) solution for the more generalk-differences pro-blems, where insertions and deletions are also considered.
The idea of the algorithm for approximate matching stems from the fact that extensions may be intercalated with mismatches. Therefore, ifkmismatches are allowed,k
+ 1 extensions may be performed to match a patternPof sizeLin a substringS[i..i+L -1], with 1≤i≤N - L+ 1. Roughly speaking, match operations are as follows: (i) per-form a LCE query to matchP[1..L] inS[i..i+L-1]; (ii) if a mismatch takes place in positionj < LofP, jump over the mismatch and go to (i) to perform another LCE query to matchP[j+ 1..L] inS[i+j+ 1..i+L+ 1]. This procedure must be undertaken recursively untilkmismatches are reached or the end ofPis reached. If the extensions does not reach the end ofPthen more thankmismatches are needed. Conversely, if the extensions reach the end ofP
then P[1..L] matches S[i..i + L - 1] with at most k
mismatches.
Concerning time complexity, each match operation takes at mostO(k) time, since at mostk+ 1 extensions are tried and each extension takes O(1). The scan over all string S takes O(N), therefore the k-mismatch
problem takes O(Nk) time. A detailed explanation, as well as the pseudo-code of the algorithm, can be found in Additional file 2. This procedure needs only the reverse CGR index ofS, without using any special index data structure.
A simple variant of thek-mismatch algorithm can be used to solve the allk-mismatch palindromes. The idea is always to perform an extension and jump over a mismatch to perform another extension until the number of allowed mismatches are attained. Consequently, the complexity of allk-mismatch palindromes is updated toO(Nk) time. The algorithm for allk-mismatch tandem repeats [46] is also coincident, although more complex, and is fully described in the Additional file 2.
Longest common substring
The retrieval of the longest common substring of two strings is a classical computer science problem. Given a string S1 of sizeN and a string S2 of sizeM, a naive approach to this problem simply consider all positionsiin
S1andjinS2and proceed in both strings by matching symbols, until a mismatch is found. Such procedure takes
O(NM2) time. By using linear time search algorithms, such as the Knuth-Morris-Pratt, this result can be improved to
O(NM). Still, the performance of these naive solutions pales in comparison with the linear time performance of suffix trees [4].
In the CGR framework we propose an algorithm that does not achieve linear time complexity, as suffix tree does, but it is sufficiently close to warrant a careful analy-sis. We start this analysis by considering the simpler pro-blem of finding duplicates in a list of numbers. Assume we are given an unordered list of numbersℓ, of sizeN, and we wish to determine if there are any repeated number. A naive solution would compare every pair of numbers to check if they are equal, requiringO(N2) time. A more sophisticated solution would be to sort the listℓand check only consecutive numbers. This algorithm requires overall
O(NlogN) time, where the sorting step is the bottleneck of the procedure.
To obtain the longest common substring from the CGR representation we join the CGR coordinates fromS1and
S2 and sort them all together, according to the previous procedure. Then, we traverse the sorted list and, if two consecutive coordinates come from two different strings, i. e., one fromS1and the other fromS2, we compute the LCE between the two. Overall our goal is to determine the pair that has the largest LCE value, as this corresponds the longest common substring among the two strings.
Rabin-Karp string matching algorithm
The procedure for calculating the CGR coordinates (Eq. (1) and (2)) bare a striking similarity to Rabin-Karp string matching algorithm [47]. This algorithm uses arolling hash function h, from strings to integers, allowing to reduce the problem of comparing strings to the problem of comparing integers, which can be computed much more efficiently in commodity hardware.
Given a stringSof sizeNand a patternPof sizeL, the Rabin-Karp algorithm starts by computing the codeh(P). Then, allL-tuples ofSare scanned in order to check ifh(S
[i..i+L- 1]) =h(P), with 1≤i≤N - L+ 1. The substrings for whichh(P) =h(S[i..i+L -1]) are potential occurrences ofP, which should be further checked naively. This algo-rithm was one of the firsts to achieve linear pattern match-ing efficiency, takmatch-ing onlyO(N) time to scan a string of sizeN. Notwithstanding, it results in a randomized algo-rithm, sinceh(P) =h(S[i..i+L -1]) does not constitute a guarantee thatP=S[i..i+L -1] and so, an additional check is needed to verify ifP=S[i..i+L -1]. In conse-quence, its performance is onlyO(N) in the expected case, and the worst case may be significantly worse, takingO
(N2) time. Other linear time algorithms such as the Knuth-Morris-Pratt and the Boyer-Moore can guarantee linear time performance, even in the worst case [35,48].
The Rabin-Karp algorithm has the advantage of produ-cing and index as a side effect, which is actually a hash. Indexes have the advantage that they support subsequent string searches in much less time than searching over the all stringS. However, their performance depend largely on the nature of the underlying hash. If the hash values of consecutive substrings are computed in constant time, it is possible to achieve almost aO(L+occ) search time, mean-ing that searchmean-ing for all the occurrencesoccof aL-long patternPcan be performed in expected optimal time [49]. This is exactly what is attained when CGR is used to induce a rolling hash function for the Rabin-Karp procedure.
In the context of CGR, a possible rolling hash function can be constructed iteratively based on the CGR defini-tion (Eq. (1)) and the LEFT DELETION operadefini-tion described in Eq. (6). If one uses directly the coordinates of the last symbol of the firstL-tuple to compute the first hash value we have that
h(S[1..L]) =xLS= (1−r)Lx0+r
L
k=1
(1−r)L−kySk.
The next hash valueh(S[2..L+1]) can be again defined as the last CGR coordinate of string S[2..L+1]. This coordinate xSL[2..L+1] can be computed from xSL[1..L]=xS
L by performing a LEFT DELETION and applying a new CGR iteration. The computation of this value is straight-forward. First, the coordinate xSL[2..−1L] is obtained by
canceling symbolS[1] from xS L, i.e.,
xSL[2..−1L]=xSL−r(1−r)L−1(yS1−x0).
Then, to obtain the intended coordinate xSL[2..L+1], a CGR iteration is applied and we get
xLS[2..L+1] = (1−r)xLS−[2..1L]+rySL+1,
which can be defined in terms of xSL as
xSL[2..L+1]= (1−r)xSL−r(1−r)L(y1S−x0) +ryLS+1.
Thus, the Rabin-Karp CGR-based procedure starts by defining h(S[1..L])=xSL and proceeds by computing iteratively the code forh(S[i+ 1..i+L]) as
h(S[i+1..i+L]) = (1−r)·h(S[i..i+L−1])−r(1−r)L
(S[i]−x0)+r·S[i+L], i= 1,. . .,N−L. (7)
Even though this expression uses vectors and is there-fore slightly more sophisticated than the most common expressions, CGR function is essentially a hash, except for the fact the we are not using remainders as in the original proposal. However, due to the limitation of the floating point representation, iterating Eq. (1) will even-tually overflow, hence producing the same effect as explicitly computing the remainders. If this occurs forh
(P) a spurious hit might be obtained, requiring a naive verification. Naturally, infinite precision techniques can be used to prevent this phenomena, which in turn yield powerful computation models.
Finally, with Eq. (7) it is possible to update the hash value in constant time and, therefore, obtaining all the hash values takes only O(N) for a string S of sizeN. Likewise a hash data structure that indexes all L-tuples ofScan be built within this time.
Conclusions
CGR has been, for more than two decades, a fruitful methodology for biological sequence comparison, provid-ing a support for aligment-free analysis along with convey-ing an appealconvey-ing pattern visualization capacity for whole genomes. In this work we have shown that CGR can go beyond these applications and demonstrate how typical string operations can be recoded and solved using CGR. We further illustrate the similarities between these two data structures for biological sequence analysis, showing that numerical biosequence representations given by chaos game iterative function systems constitute an alter-native and competitive approach for common string-matching problems.
The applications presented in this work have focused on the longest common extension problem that, after linear time preprocessing of a standard CGR index, it is shown to be answered in constant time. This result allows to efficiently solve other string matching pro-blems such as searching for palindromes, tandem repeats and matches with mismatches. Additionally, it is shown that CGR can be used as an hash function and its relation with Rarbin-Karp algorithm is highlighted.
The chief advantage of CGR is its simplicity and easy implementation. In addition, a more complex placement of the CGR coordinates in memory actually provides an efficient way to solve more demanding problems, for example, through the use of quadtrees, here proposed to solve the longest common substring problem.
The choice between numerical or graphical resolution is ultimately decided by the efficiency of the implemen-tation. To assist in this selection we provide a summary of time complexities achieve by both data structures for a set of pertinent problems in string processing for molecular biology, showing the CGR parallelism with suffix trees in some of the problems addressed.
The operations analyzed covered typical string problems without being exhaustive. Possible extensions to be explored numerically include sequence alignment, which can be implemented using a dynamic programming approach in a matrix of all the pairwise distances between the CGR coordinates. The longest common subsequence can also be addressed in the future using numerical reasoning.
The comparison of CGR and suffix trees algorithms for biological sequence analysis provides a useful bridge between graph and numerical-based formalisms such that solutions to difficult problems can make use of both representations.
Additional material
Additional file 1: Longest common extension queries. Additional file 1 presents in detail the problem of computing LCE queries with CGR
maps that uses a contraction parameter ratio ofr= 1
2. It also presents
the solution by embedding the CGR coordinates in the Cantor set and
shows that this corresponds to the CGR map withr= 2 3.
Additional file 2: Detailed algorithms. Additional file 2 presents in detail the algorithms sketched throughout the section that exploits LCE queries in constant time.
Acknowledgements
This work was partially supported by FCT through the PIDDAC Program funds (INESC-ID multiannual funding) and under grant PEst-OE/EEI/LA0008/ 2011 (IT multiannual funding). In addition, it was also partially funded by projects HIVCONTROL (PTDC/EEA-CRO/100128/2008, S. Vinga, PI), TAGS (PTDC/EIA-EIA/112283/2009) and NEUROCLINOMICS (PTDC/EIA-EIA/111239/ 2009) from FCT (Portugal).
Author details
1Instituto de Engenharia de Sistemas e Computadores: Investigação e
Desenvolvimento (INESC-ID), R. Alves Redol 9, 1000-029 Lisboa, Portugal.
2Dept Bioestatística e Informática, Faculdade de Ciências Médicas
-Universidade Nova de Lisboa (FCM/UNL), C. Mártires Pátria 130, 1169-056 Lisboa, Portugal.3Instituto de Telecomunicações (IT), Av. Rovisco Pais 1, Torre
Norte, Piso 10, 1049-001 Lisboa, Portugal.4Instituto Superior Técnico, Universidade Técnica de Lisboa (IST/UTL), Av. Rovisco Pais 1, 1049-001 Lisboa, Portugal.5Div Informatics, Dept Pathology, University of Alabama at Birmingham, USA.
Authors’contributions
SV conceived the study and contributed for the design of the experiments. AMC aimed the research to achieve longest common extension in constant time with standard CGR indexes. APF implemented the JavaScript version and analyzed the different implementations. LR analyzed the CGR against string processing algorithm such as suffix trees and Rabin-Karp. JSA identified programming environment and contributed to the discussion of results. All authors contributed to the writing of the manuscript. All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Received: 25 January 2012 Accepted: 2 May 2012 Published: 2 May 2012
References
1. Durbin R, Eddy SR, Krogh A, Mitchison G:Biological sequence analysis:
probabalistic models of proteins and nucleic acidsCambridge, UK New York:
Cambridge University Press; 1998.
2. Shendure J, Ji H:Next-generation DNA sequencing.Nature Biotechnology
2008,26(10):1135-1145.
3. Roy A, Raychaudhury C, Nandy A:Novel techniques of graphical representation and analysis of DNA sequences - A review.Journal of Biosciences1998,23:55-71.
4. Gusfield D:Algorithms on strings, trees, and sequences: computer science and
computational biologyCambridge England; New York: Cambridge University
Press; 1997.
5. Navarro G, Mäkinen V:Compressed Full-Text Indexes.ACM Computing Surveys2007,39, article 2.
6. Langmead B, Trapnell C, Pop M, Salzberg S:Ultrafast and memory-efficient alignment of short DNA sequences to the human genome.Genome Biol
2009,10(3):R25.
7. Li H, Durbin R:Fast and accurate short read alignment with Burrows-Wheeler transform.Bioinformatics2009,25(14):1754-1760.
8. Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K, Wang J:SOAP2: an improved ultrafast tool for short read alignment.Bioinformatics2009, 25(15):1966-1967.
9. Fernandes F, da Fonseca PGS, Russo LMS, Oliveira AL, Freitas AT:Efficient alignment of pyrosequencing reads for re-sequencing applications.BMC Bioinformatics2011,12:163.
10. Jeffrey HJ:Chaos game representation of gene structure.Nucleic Acids Res
11. Arakawa K, Oshita K, Tomita M:A web server for interactive and zoomable Chaos Game Representation images.Source code for biology and medicine2009,4:6.
12. Deschavanne PJ, Giron A, Vilain J, Fagot G, Fertil B:Genomic signature: characterization and classification of species assessed by chaos game representation of sequences.Mol Biol Evol1999,16(10):1391-9. 13. Karlin S, Burge C:Dinucleotide relative abundance extremes: a genomic
signature.Trends Genet1995,11(7):283-90.
14. Wang YW, Hill K, Singh S, Kari L:The spectrum of genomic signatures: from dinucleotides to chaos game representation.Gene2005, 346:173-185.
15. Almeida JS, Carrico JA, Maretzek A, Noble PA, Fletcher M:Analysis of genomic sequences by Chaos Game Representation.Bioinformatics2001, 17(5):429-37.
16. Vinga S, Almeida J:Alignment-free sequence comparison-a review.
Bioinformatics2003,19(4):513-23.
17. Vinga S, Almeida JS:Renyi continuous entropy of DNA sequences.J Theor Biol2004,231(3):377-88.
18. Vinga S, Almeida JS:Local Renyi entropic profiles of DNA sequences.BMC Bioinformatics2007,8:393.
19. Pandit A, Sinha S:Using genomic signatures for HIV-1 sub-typing.BMC Bioinformatics2010,11(Suppl 1):S26.
20. Rasouli M, Rasouli G, Lenz FA, Borrett DS, Verhagen L, Kwan HC:Chaos game representation of human pallidal spike trains.J Biol Phys2010, 36(2):197-205.
21. Dufraigne C, Fertil B, Lespinats S, Giron A, Deschavanne P:Detection and characterization of horizontal transfers in prokaryotes using genomic signature.Nucleic Acids Research2005,33:e6.
22. Deschavanne P, Tuffery P:Exploring an alignment free approach for protein classification and structural class prediction.Biochimie2008, 90(4):615-625.
23. Almeida JS, Vinga S:Computing distribution of scale independent motifs in biological sequences.Algorithms for molecular biology: AMB2006,1:18. 24. Almeida JS, Vinga S:Universal sequence map (USM) of arbitrary discrete
sequences.BMC Bioinformatics2002,3:6.
25. Almeida J, Vinga S:Biological sequences as pictures - a generic two dimensional solution for iterated maps.BMC Bioinformatics2009,10:100. 26. Buhlmann P, Wyner A:Variable length Markov chains.Annals of Statistics
1999,27(2):480-513.
27. Tino P, Dorffner G, Schittenkopf C:Understanding state space organization in recurrent neural networks with iterative function systems dynamics.InHybrid Neural Systems, Volume 1778 of Lecture Notes in Artificial IntelligenceEdited by: Wermter S, Sun R 2000, 255-269, [International Workshop on Hybrid Neural Systems, DENVER, CO, DEC 04-05, 1998].
28. Tino P, Dorffner G:Predicting the future of discrete sequences from fractal representations of the past.Machine Learning2001,45(2):187-217. 29. Moreno P, Velez P, Martinez E, Garreta L, Diaz N, Amador S, Tischer I,
Gutierrez J, Naik A, Tobar F, Garcia F:The human genome: a multifractal analysis.BMC Genomics2011,12:506.
30. Ferreirós J:Labyrinth of Thought: A History of Set Theory and Its Role in
Modern MathematicsBirkhäuser Basel; 2007.
31. Willard S:General TopologyDover Publications; 2004.
32. Samet H:The quadtree and related hierarchical data-structures.
Computing Surveys1984,16(2):187-260.
33. Cenac P, Chauvin B, Ginouillac S, Pouyanne N:Digital Search Trees And Chaos Game Representation.ESAIM-Probability and Statistics2009, 13:15-37.
34. Ukkonen E:On-line construction of suffix trees.Algorithmica1995, 14(3):249-260.
35. Knuth D, Morris J Jr, Pratt V:Fast pattern matching in strings.SIAM Journal
on Computing1977,6(2):323-350.
36. Aho A, Corasick M:Efficient string matching: an aid to bibliographic search.Communications of the ACM1975,18(6):333-340.
37. Pingoud A, Jeltsch A:Structure and function of type II restriction endonucleases.Nucleic Acids Research2001,29(18):3705-3727. 38. Reich NO, Danzitz MJ:Non-additivity of sequence-specific enzyme-DNA
interactions in the EcoRI DNA methyltransferase.Nucleic Acids Research
1991,19(23):6587-6594.
39. Ussery DW, Wassenaar T, Borini S:Word Frequencies, Repeats, and Repeat-related Structures in Bacterial Genomes. Springer2008.
40. Kidwell MG:Horizontal transfer of P elements and other short inverted repeat transposons.Genetica1992,86(1-3):275-286.
41. Vinces MD, Legendre M, Caldara M, Hagihara M, Verstrepen KJ:Unstable Tandem Repeats in Promoters Confer Transcriptional Evolvability.Science
2009,324(5931):1213-1216.
42. Baumstark AL, Budowle B, Defenbaugh DA, Smerick JB, Keys KM, Moretti TR: Validation of Short Tandem Repeats (STRs) for Forensic Usage: Performance Testing of Fluorescent Multiplex STR Systems and Analysis of Authentic and Simulated Forensic Samples.Journal of Forensic Sciences
1999,46(3):647-660.
43. Landau GM, Schmidt JP:An Algorithm for Approximate Tandem Repeats.
CPM, Volume 684 of LNCS1993, 120-133.
44. Landau GM, Vishkin U:Introducing Efficient Parallelism into Approximate String Matching and a New Serial Algorithm.InSTOC.Edited by: Hartmanis J. ACM; 1986:220-230.
45. Myers E:An O(ND) Difference Algorithm and Its Variations.Algorithmica
1986,1(2):251-266.
46. Landau GM, Schmidt JP, Sokol D:An Algorithm for Approximate Tandem Repeats.Journal of Computational Biology2001,8:1-18.
47. Karp R, Rabin M:Efficient randomized pattern-matching algorithms.IBM
Journal of Research and Development1987,31(2):249-260.
48. Cole R:Tight bounds on the complexity of the Boyer-Moore string matching algorithm.Proceedings of the second annual ACM-SIAM
symposium on Discrete algorithmsSODA‘91, Philadelphia, PA, USA: Society
for Industrial and Applied Mathematics; 1991, 224-233.
49. Cormen TH, Leiserson CE, Rivest RL, Stein C:Introduction to algorithmsMIT Press; 2009.
doi:10.1186/1748-7188-7-10
Cite this article as:Vingaet al.:Pattern matching through Chaos Game Representation: bridging numerical and discrete data structures for biological sequence analysis.Algorithms for Molecular Biology20127:10.
Submit your next manuscript to BioMed Central and take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution