Hypotonia and Lethargy in
a Two-Day-Old Male Infant
Adrienne H. Long, MD, PhD,a,bJennifer G. Fiore, MD,a,bRiaz Gillani, MD,a,bLaurie M. Douglass, MD,cAlan M. Fujii, MD,d Jodi D. Hoffman, MDe
abstract
A 2-day old term male infant was found to be hypotonic and minimallyreactive during routine nursing care in the newborn nursery. At 40 hours of life, he was hypoglycemic and had intermittent desaturations to 70%. His mother had an unremarkable pregnancy and spontaneous vaginal delivery. The mother’s prenatal serology results were negative for infectious risk factors. Apgar scores were 9 at 1 and 5 minutes of life. On day 1 of life, he fed, stooled, and voided well. Our expert panel discusses the differential diagnosis of hypotonia in a neonate, offers diagnostic and management
recommendations, and discusses thefinal diagnosis.
DRS LONG, FIORE, AND GILLANI, PEDIATRIC RESIDENTS
A 2-day old male infant born at 38 weeks and 4 days was found to be limp and minimally reactive during routine care in the newborn nursery. Just 5 hours before, he had an appropriate neurologic status when evaluated by the same nurse. He had breastfed once in the interim.
His mother received routine prenatal care in the United States without complications. Prenatal ultrasounds and maternal prenatal serologies were unremarkable. His mother had a history of a positive purified protein derivative tuberculin test and was treated for latent tuberculosis 3 years before. She had no other medical history. The parents immigrated from El Salvador 1 year before and had not traveled out of the country since that time. The couple’sfirst child was a girl who died in her sleep in El Salvador on the second day of life of unknown cause. The parents denied consanguinity.
The patient was born via spontaneous vaginal delivery to a now gravida 2, para 2 mother with Apgar scores of 9 and 9 at 1 and 5 minutes of life. His
birth weight was 3.4 kg (56th percentile), length was 52 cm (87th percentile), and head circumference was 33 cm (12th percentile). His physical examination at birth was normal for gestational age, with appropriate neurologic, cardiac, and respiratory components. The infant uneventfully roomed with his mother during thefirst 2 days of life. He breastfed well every 2 to 3 hours, with good latching during lactation
assessments. He appropriately stooled and voided. Routine physical
assessments were performed every 8 hours per protocol, which revealed a consistently normal physical examination including neurologic status. The newfinding on day of life 2 that he was limp and minimally reactive represented a significant change from his previous clinical status. The pediatric resident on call presented to evaluate the patient. The infant was found ill appearing with a temperature of 35.4°C, pulse of 110 beats per minute, respiratory rate of 55 beats per minute, and blood pressure of 92/50 mm Hg. Oxygen saturation was 94% on room air with intermittent brief self-resolving desaturations to 70%. He was
aDepartment of Medicine, Boston Children’s Hospital, Boston, Massachusetts; anddNeonatology Section,eMedical Genetics Section,cDivision of Child Neurology, and bDepartment of Pediatrics, Boston Medical Center, Boston, Massachusetts
Drs Long, Fiore, and Gillani conceptualized, drafted, and edited the manuscript; Drs Douglass, Fujii, and Hoffman edited the manuscript; and all authors approved thefinal manuscript as submitted and agree to be accountable for all aspects of the work.
DOI:https://doi.org/10.1542/peds.2018-0788
Accepted for publication Dec 18, 2018
Address correspondence to Adrienne H. Long, MD, PhD, Department of Pediatrics, Boston Children’s Hospital, 300 Longwood Ave, Hunnewell Building, Pavilion 129, Housestaff Lounge, Boston, MA 02115. E-mail: adrienne.long@childrens.harvard.edu
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online, 1098-4275).
Copyright © 2019 by the American Academy of Pediatrics
FINANCIAL DISCLOSURE:The authors have indicated they have nofinancial relationships relevant to this article to disclose.
FUNDING:No external funding.
POTENTIAL CONFLICT OF INTEREST:The authors have indicated they have no potential conflicts of interest to disclose.
sleepy with a weak cry. He was normocephalic with no distinctive features. On pulmonary examination, he had mild supraclavicular and subcostal retractions with good air entry bilaterally. He was diffusely hypotonic with a prominent head lag. The Moro reflex was absent. He had a good suck reflex. Palmar grasp and plantar grasp were present but weak. Muscle bulk was normal. He had normal cardiac, abdominal, dermatologic, and genitourinary examinations. He had strong radial and femoral pulses and was well perfused throughout.
HOW WOULD YOU APPROACH THE INITIAL MANAGEMENT OF THIS NEWBORN WITH HYPOTONIA, HYPOTHERMIA, AND DESATURATIONS IN THE NICU? WHAT INITIAL
EVALUATION IS WARRANTED?
Dr Fujii, Neonatology
The constellation of symptoms, including hypotonia, hypothermia, and desaturations, is alarming for a serious process in this 2-day-old infant. Although a broad differential should be considered, the acuity and timing of the presentation can narrow the differential to a smaller set of more likely diagnoses. For example, had the patient developed symptoms immediately after birth, hypoxic ischemic encephalopathy (HIE) would be high on the differential. However, it would be unlikely for HIE to manifest acutely after 48 hours of otherwise normal activity. On the basis of pathophysiology and epidemiology, hypoglycemia or sepsis would be high on my differential.
Severe hypoglycemia itself can lead to a presentation similar to this infant’s, with altered mental status, poor tone, and an inability to maintain body temperature. Desaturations in this setting could be explained by hypoglycemic seizures. In term infants, common risk factors for hypoglycemia include being small for gestational age, large for gestational
age, being an infant of a mother with diabetes, or having a maternal history ofb-blocker usage.1,2This patient did not have these risk factors. Although the absence of such risk factors is less common, it does not exclude the possibility that hypoglycemia was the instigating factor that led to the presentation.
Neonatal sepsis can also present similarly. Indications of neonatal sepsis are often nonspecific and include poor feeding, lethargy, temperature instability (including hypothermia), and respiratory symptoms. Furthermore, sepsis can lead to hypoglycemia through poor feeding and increased metabolic demands. Although this patient does not have risk factors associated with early neonatal sepsis, namely positive maternal group B streptococcus (GBS) status, prolonged rupture of membranes, or elevated maternal intrapartum temperature, sepsis should still be high on the
differential.3Neonatal sepsis occurs in 1 in 1000 births and causes 15% of all neonatal deaths.4–6Furthermore, although GBS infections remain the leading cause of early-onset neonatal sepsis (∼36%),7∼80% of GBS infections occur in patients whose mothers had negative perinatal test results.8Thus, this patient warrants complete workup for sepsis and empirical treatment.
In the general population, other diagnoses beyond these are significantly rarer. Such diagnoses could include in utero insults, neuromuscular disorders, metabolic and/or genetic disorders,
endocrinological issues, and congenital heart disease.9However, such diagnoses should remain higher on the differential in patients such as this who have a positive family history of infant death.
Initial management should be focused on stabilizing the infant and
evaluating the most likely causes of illness. I would start with evaluation
for hypoglycemia and sepsis and provide respiratory support. I would obtain a point-of-care glucose, complete metabolic panel, complete blood count (CBC) with differential, blood cultures, and a lumbar puncture with cerebrospinalfluid (CSF) studies and culture. Urine culture in this setting is of low utility because isolated urinary tract infections are exceptionally
uncommon in infants,1 week old.10 I would empirically start broad-spectrum antibiotics. Regarding the patient’s desaturations and increased work of breathing, I would obtain a chest radiograph and venous blood gas. I would tailor respiratory support to the degree of respiratory distress.
Drs Long, Fiore, and Gillani
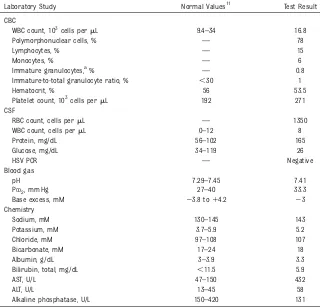
The infant was transferred to the NICU for further management. Laboratory studies, including a full sepsis workup with lumbar puncture, were obtained (Table 1). Point-of-care glucose was 32 mg/dL. The CBC was reassuring, with a normal white blood cell count for age (16.83103 cells/mL) and normal immature-to-total granulocyte ratio (1%). The CSF cell count was consistent with a mildly traumatic tap without significant pleocytosis. CSF
chemistries revealed a low glucose of 26 mg/dL, consistent with systemic hypoglycemia and a mildly elevated protein of 165 mg/dL. Serum chemistry was normal except for an isolated elevation in aspartate aminotransferase (AST) (432 U/L) with a high-to-normal alanine aminotransferase (ALT) (58 U/L). Venous blood gas from the infant was unremarkable. His chest radiograph was normal.
infant’s clinical status significantly improved, though did not completely normalize. His lethargy, hypothermia, and hypoglycemia completely resolved. He had no additional hypoxic episodes and the increased work of breathing resolved. The diffuse hypotonia observed on previous examinations was improved, although he still had a slightly increased head lag, incomplete Moro reflex, slightly increased deep tendon reflexes (DTRs), and mildly weak palmar grasp. There was no evidence of muscle fasciculations.
The infant’s rapid clinical
improvement with the normalization of his serum glucose strongly suggested hypoglycemia contributed to, at least in part, his initial decompensation. The rapid timeline for improvement was also thought to be less consistent with sepsis for an underlying etiology, especially in the
setting of a normal CBC. However, an infectious etiology could not be ruled out; thus, the patient remained on empirical antibiotics. Importantly, the infant’s persistent, albeit mild, abnormalities on neurologic examination in the setting of euglycemia prompted consideration of additional underlying causes that led to his acute decompensation, such as neurologic and metabolic
etiologies.
WHAT WOULD BE THE NEUROLOGIC AND METABOLIC DIFFERENTIAL DIAGNOSIS FOR THIS PATIENT?
Dr Douglass, Neurology
The differential diagnosis for a newborn with hypotonia is broad. The localization can be anywhere along the neural axis: the brain, spinal cord, nerve, neuromuscular junction, or muscle.12However, identifying
central versus peripheral hypotonia can help narrow the differential. To do this, it is important to determine if the infant has hypotonia and/or weakness.13Hypotonia refers to decreased resistance to stretch, whereas weakness refers to decreased muscle strength. Central hypotonia is accompanied by increased DTRs and often altered mental status. Weakness with central hypotonia is less apparent at this age because the newborn’s movements are less controlled by the cortex. Examples of central etiologies include structural abnormalities such as cerebral malformation, HIE, and stroke. In contrast, peripheral hypotonia is associated with noticeable weakness, which may present as a weak cry, a weak grasp, a reduced Moro response, or decreased antigravity movements. These infants are often more alert. Examples for peripheral hypotonia include spinal muscular atrophy, congenital muscular dystrophies, and congenital or metabolic myopathies. Overall, central hypotonia is significantly more common than peripheral hypotonia in neonates.
After resolution of his hypoglycemia, this patient had persistent neurologic findings consistent with both central (increased DTRs) and peripheral hypotonia (mild weakness and normal mental status). In this setting, one should consider mixed muscle-brain etiologies, such as congenital muscular dystrophies or metabolic disorders.
Timing of when the infant developed hypotonia also provides important information. The acute change in his tone makes cerebral dysgenesis, syndromic causes such as Down syndrome or Prader-Willi syndrome, or spinal muscular atrophy less likely. The most common causes of sudden onset hypotonia in a newborn are toxic, metabolic, or infectious. Muscular conditions tend to have an insidious onset.
TABLE 1Initial Laboratory Evaluation (Day of Life 2)
Laboratory Study Normal Values11 Test Result
CBC
WBC count, 103cells permL 9.4–34 16.8
Polymorphonuclear cells, % — 78
Lymphocytes, % — 15
Monocytes, % — 6
Immature granulocytes,a% — 0.8
Immature-to-total granulocyte ratio, % ,30 1
Hematocrit, % 56 53.5
Platelet count, 103cells permL 192 271
CSF
RBC count, cells permL — 1350
WBC count, cells permL 0–12 8
Protein, mg/dL 56–102 165
Glucose, mg/dL 34–119 26
HSV PCR — Negative
Blood gas
pH 7.29–7.45 7.41
PCO2, mm Hg 27–40 33.3
Base excess, mM 23.8 to14.2 23
Chemistry
Sodium, mM 130–145 143
Potassium, mM 3.7–5.9 5.2
Chloride, mM 97–108 107
Bicarbonate, mM 17–24 18
Albumin, g/dL 3–3.9 3.3
Bilirubin, total, mg/dL ,11.5 5.9
AST, U/L 47–150 432
ALT, U/L 13–45 58
Alkaline phosphatase, U/L 150–420 131
HSV, herpes simplex virus; PCR, polymerase chain reaction; RBC, red blood cell; WBC, white blood cell;—, not applicable.
Dr Hoffman, Genetics
Syndromic causes of hypotonia are important to explore. Prader-Willi syndrome can present as hypotonia and poor feeding in a newborn without overt physical signs of a genetic syndrome,14although I agree that the patient’s acute decompensation may make such syndromes less likely. Metabolic disorders can certainly cause hypotonia and hypoglycemia in a neonate. Thinking about metabolic causes, I like the approach outlined by Saudubray et al,15which identifies 3 broad categories: disorders of intoxication, disorders of energy metabolism, and disorders of complex molecules.
Disorders of intoxication are due to problems with the use of basic nutrients and include amino acid and urea cycle disorders, organic
acidemias, and disorders of metal intoxication. The toxic metabolites resulting from these disorders can lead to a worsening clinical picture over time. Disorders of energy metabolism are due to problems with energy production and use; examples include mitochondrial disorders, disorders of gluconeogenesis and glycogen storage, pyruvate
metabolism disorders, and disorders of fatty acid oxidation. Finally, disorders of complex molecules include entities such as lysosomal and peroxisomal disorders; these tend to be rarer overall.
In the short-term, laboratories that would help explore the broad differential under inborn errors of metabolism are urinalysis for assessment of urine ketones, creatine kinase (CK), ammonia, and lactate. Blood amino acids, acylcarnitines, urine organic acids, and acylglycines would be helpful, but results would not be available immediately.
Dr Fujii
State newborn screenings are also extremely useful in these
circumstances. Such tests have high sensitivity and are excellent screening studies for many inherited metabolic diseases. In many states, expedited testing can be requested for infants suspected of having metabolic disorders.
Drs Long, Fiore, and Gillani
Given concerns for possible neurologic or metabolic etiologies, further imaging and blood studies were pursued. A head ultrasound was unremarkable. Metabolic laboratories were drawn in the context of resolving hypoglycemia (Tables 2 and 3). These studies were notable for a significant elevation in CK to.38 000 U/L (upper limit of normal for age is ∼800 U/L). Urinalysis showed trace ketones. Ammonia, lactate, and thyroid studies were normal. An expedited New England Newborn Screen was sent. Plasma and urine amino acids, urine organic acids, plasma
acylcarnitines, and urine acylglycines were sent and were anticipated to take several days before the results would be known.
In addition to the significantly elevated CK, recall that the infant had an isolated elevation in AST without a similar increase in ALT (Table 1). Bagged urinalysis revealed 31blood on dipstick without an increase in red
blood cells on microscopic examination (Table 2). Together, these studies were highly suggestive of significant rhabdomyolysis. Further assessment of cardiac involvement revealed a mild increase in the CK–muscle-brain isoform; however, this was thought to be most likely cross-reactivity in the setting of significantly elevated total CK levels (cardiac index 0.5%) (Table 3). Reassuringly, there was no associated increase in troponin I, and an echocardiogram was normal. Renal function did not appear significantly impaired because the infant’s creatinine was not elevated beyond what would be expected 48 hours after birth.
HOW DO THE ADDITIONAL STUDIES NARROW THE DIFFERENTIAL?
Dr Douglass
Given the degree of CK elevation, a congenital muscular dystrophy comes to the top of the neurologic differential. These can have related central nervous system
malformations and can be consistent with this patient’s mixed peripheral/ central hypotonia. Pure central nervous system diagnoses are much less likely at this point. Inborn errors of metabolism, however, are also on the differential.
TABLE 2Metabolic Laboratory Evaluation
Laboratory Study Normal Values11 Test Result
Serum metabolic studies
Glucose, mg/dL 50–90 80
Lactate, mM 1.1–3.5 2.1
Ammonia,mM 56–92 49
CK, U/L 145–1578 .38 000
Thyroid stimulating hormone,mIU/mL 2.43–24.3 3.27
T4, ng/dL 0.94–4.39 1.16
Urinalysis (bag specimen)
Protein Negative 21
Ketone Negative Trace
Blood Negative 31
Leukocyte esterase Negative Negative
Bacteria Negative Positive
RBC count, cells per HPF 0–3 0–3
WBC count, cells per HPF 0–5 6–10
Dr Hoffman
With regard to the inborn errors of metabolism, the patient appears relatively hypoketotic. It is important to note that urine ketones can be challenging to interpret in the newborn period because many infants poorly produce ketones. However, this patient’s low ketones in the setting of hypoglycemia and significant rhabdomyolysis moves metabolic disorders such as fatty acid oxidation disorders much higher on the differential. However, this degree of rhabdomyolysis in the early newborn period would be an atypical presentation. Severe rhabdomyolysis typically is a dominant feature of fatty acid oxidation disorders that present in adolescence rather than in infancy. Although metabolic causes are now high on the differential, additional testing is necessary to make thefinal diagnosis.
HOW DO YOU ADDRESS THE SAFETY OF FEEDING AN INFANT WHEN METABOLIC DISORDERS ARE ON THE DIFFERENTIAL?
Dr Hoffman
When an inborn error of metabolism is suspected, caution with feeding is necessary. One must be mindful that in disorders of intoxication, toxic metabolites upstream of the blocked metabolic pathway can impair organ function. Thus, it is critical to remove the nutrient suspected to be
precipitating the illness. In the case of suspected fatty acid oxidation disorders, it is imperative to remove fats from the diet and provide the patient with glucose as an immediate source of energy. Ongoing hydration
for renal protection in this patient with significant rhabdomyolysis is also critical.
Drs Long, Fiore, and Gillani
Given concern for a fatty acid oxidation disorder, breast milk and formula were restricted to eliminate fat from the diet, pending results of the metabolic workup. The patient was maintained on dextrose
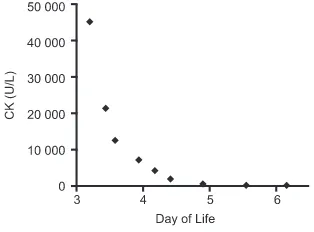
containing intravenousfluids. The CK substantially downtrended with these measures (Fig 1), which suggested that an underlying metabolic disorder (in which muscle damage was abrogated by limiting feeds) may be more likely than a muscular
dystrophy. Throughout this time, the infant continued to be well appearing, with only mildly persistent hypotonia and slightly increased DTRs. There were no additional episodes of lethargy, profound hypotonia, hypothermia, or hypoglycemia. On day of life 4, the New England Newborn Screen resulted with an increase in very long-chain
acylcarnitine species concerning for
very long-chain acyl coenzyme A dehydrogenase deficiency (VLCADD), a fatty acid oxidation disorder as suspected above. Confirmation plasma acylcarnitine profile (Fig 2) revealed marked elevation in long and very long-chain acylcarnitine species (C12-C18), including tetradecenoylcarnitine (C14:1), the most characteristic metabolic marker of VLCADD. Ultimately, genetic sequencing confirmed the VLCADD diagnosis by identifying mutations in both alleles of theACADVLgene.
FINAL DIAGNOSIS
VLCADD is a rare autosomal recessive condition caused by mutations in the
ACADVLgene, resulting in
a deficiency in the rate-limiting step of fatty acidb-oxidation for long-chain fatty acids.16The prevalence is reported to be 1 in 30 000 to 100 000 people.17Skeletal muscle and cardiac muscle rely on fatty acidb-oxidation for energy. During a fasting state when glycogen stores are depleted, patients with VLCADD cannot mobilize energy from fat stores, leading to hypoketotic hypoglycemia, lethargy, and muscle damage. Buildup of toxic long-chain fatty acid
precursors leads to multiorgan damage affecting the liver, heart, and skeletal muscle.
There are 3 typical phenotypes described in patients with VLCADD.18The most severe form presents in early infancy and is characterized by hypoglycemia, hypotonia, cardiomyopathy, arrhythmias, and hepatomegaly. Patients with this severe early-onset presentation are at risk for
multiorgan failure and fatal arrhythmias. The second form is characterized by hypoketotic hypoglycemia and hepatomegaly, without cardiac involvement, and typically presents in early childhood. The third form is characterized by exercise intolerance and
rhabdomyolysis without TABLE 3Evaluation of Rhabdomyolysis
Laboratory Study Normal Values11 Test Result
CK, U/L 145–1578 45 197
CK–muscle-brain, ng/mL — 235
Cardiac index, % 2.5 0.5
Troponin I, ng/mL ,4.8 0.027
Urea nitrogen, mg/dL 2–19 16
Creatinine, mg/dL 0.3–1.0 0.6
—, not applicable.
FIGURE 1
hypoglycemia and can present in later childhood or even adulthood. Long-term prognosis is closely tied to the severity of the initial presentation and subsequent metabolic
decompensations. Patients with the severe early-onset form of VLCADD have the poorest prognosis because of the risk for fatal cardiac
manifestations.
Patients with all forms of VLCADD are counseled to adhere to a diet low in long-chain triglycerides (LCTs), avoid prolonged fasting, and avoid
dehydration to prevent complications such as symptomatic hypoglycemia, metabolic acidosis, and
rhabdomyolysis.19Even mild childhood viral illnesses that lead to diminished enteral intake can trigger catabolism in VLCADD patients. If this is suspected, serum glucose,
chemistries, liver function tests, CK, and an electrocardiogram should be obtained. Concern for a catabolic state should also prompt hydration with high-dextrosefluids (eg, dextrose 10% water with normal saline at 1.5 times maintenance) and formula feeds with a high percentage of medium-chain triglycerides (MCTs). Use of a nasogastric tube or
gastrostomy tube can be considered to assist with the management of minor illnesses. All care should be provided in consultation with a metabolism specialist.
The patient featured in this case is unique in that he had a severe and early clinical presentation with significant rhabdomyolysis yet did not exhibit any cardiac involvement. His initial presentation of hypotonia, hypoglycemia, and intermittent desaturations appropriately raised concern for sepsis or symptomatic hypoglycemia. The family history of infant death and persistent
neurologic abnormalities were important pieces of the history and physical examination that triggered prompt workup for inherited explanations of hypotonia. Although an uncommon presentation in neonates, the identification of rhabdomyolysis ultimately allowed the differential diagnosis to be narrowed, enabling the team to reach thefinal diagnosis of VLCADD and provide appropriate management. It is interesting to note, however, that VLCADD is not commonly associated with increased DTRs, which added to the complexity of this case. It is
possible that built-up toxic
metabolites contributed to both the rhabdomyolysis and central deficits in this patient.
The patient from this case remained in the NICU until day of life 10. After diagnosis of his underlying metabolic disorder, appropriate enteral feeds (low in LCT and high in MCT) were restarted to provide complete nutrition required for growth and development. He was ultimately advanced to a regimen of 50% breast milk and 50% formula rich in MCT. His CK levels continued to
downtrend. The infant had
a transient, mild increase in his serum troponin T which self-resolved. Serial echocardiograms revealed no cardiac dysfunction throughout his
hospitalization. His hypotonia fully resolved. He was discharged from the hospital with a strict feeding regimen (low in LCT and high in MCT), explicit instructions for when to seek medical care, and scheduled metabolism follow-up.
The patient was subsequently admitted to the hospital 4 times in his first 8 months of life for viral illnesses resulting in diminished oral intake and elevated CK levels. He has FIGURE 2
otherwise been growing well and meeting developmental milestones. He continues to be managed closely by a cardiologist and a metabolism/ genetics specialist. He has not developed signs of cardiomyopathy. The family received genetic
counseling; because VLCADD has an autosomal recessive inheritance pattern, the parents were advised that future offspring would have a 25% chance of being affected with VLCADD and a 50% chance of being asymptomatic carriers.
CONCLUSIONS
Evaluating an infant with hypoglycemia or hypotonia is not uncommon in the newborn nursery. For a vast majority of these cases, inherited metabolic disorders are not the underlying cause. Other etiologies such as transient hyperinsulinism, sepsis, or HIE are significantly more likely. However, as illustrated in this case, metabolic disorders should remain on the differential, especially in infants with a family history of infant death or in term infants with severe, persistent, or otherwise unexplained hypoglycemia.
Metabolic disorders canfirst present at any age, indicative of the large diversity of pathophysiology and severity that exists. Most infants who decline after an initial period of health often present with unexplained hypoglycemia, acid-base disorders, neurologic deterioration,
cardiomyopathy and/or arrhythmias, or acute parenchymal liver disease.20 Presence of thesefindings should prompt additional workup.
Beyond the initial diagnostic evaluation, criticalfirst steps are required for the management of infants with a suspected metabolic disorder. In general, this consists of minimizing catabolism and removing the toxic nutrients from the diet. These measures can be started before a precise diagnosis is made because
clinical outcomes are linked with how fast treatment is initiated.
Workup and management of infants suspected of metabolic disorders should be performed in collaboration with metabolic specialists. Multiple online resources also exist that can assist clinicians in the diagnosis and treatment of such patients. These include the New England Consortium of Metabolic Programs,21
GeneReviews,22Inborn Errors of Metabolism Knowledgebase,23and the American College of Medical Genetics and Genomics ACTion Sheets and Confirmatory Algorithms.24
ABBREVIATIONS
ALT: alanine aminotransferase AST: aspartate aminotransferase CBC: complete blood count CK: creatine kinase CSF: cerebrospinalfluid DTR: deep tendon reflex GBS: group B streptococcus HIE: hypoxic ischemic
encephalopathy LCT: long-chain triglyceride MCT: medium-chain triglyceride VLCADD: very long-chain acyl
coenzyme A dehydrogenase deficiency
REFERENCES
1. Adamkin DH; Committee on Fetus and Newborn. Postnatal glucose
homeostasis in late-preterm and term infants.Pediatrics. 2011;127(3):575–579
2. Bateman BT, Patorno E, Desai RJ, et al. Late pregnancybblocker exposure and risks of neonatal hypoglycemia and bradycardia.Pediatrics. 2016;138(3): e20160731
3. Puopolo KM, Draper D, Wi S, et al. Estimating the probability of neonatal early-onset infection on the basis of maternal risk factors.Pediatrics. 2011; 128(5). Available at: www.pediatrics. org/cgi/content/full/128/5/e1155
4. Bailit JL, Gregory KD, Reddy UM, et al. Maternal and neonatal outcomes by
labor onset type and gestational age.
Am J Obstet Gynecol. 2010;202(3): 245.e1–245.e12
5. Weston EJ, Pondo T, Lewis MM, et al. The burden of invasive early-onset neonatal sepsis in the United States, 2005-2008.
Pediatr Infect Dis J. 2011;30(11): 937–941
6. Oza S, Lawn JE, Hogan DR, Mathers C, Cousens SN. Neonatal cause-of-death estimates for the early and late neonatal periods for 194 countries: 2000-2013.Bull World Health Organ. 2015;93(1):19–28
7. Schrag SJ, Farley MM, Petit S, et al. Epidemiology of invasive early-onset neonatal sepsis, 2005 to 2014.
Pediatrics. 2016;138(6):e20162013
8. Stoll BJ, Hansen NI, Sánchez PJ, et al; Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Early onset neonatal sepsis: the burden of group B Streptococcal and E. coli disease continues.
Pediatrics. 2011;127(5):817–826
9. Leyenaar J, Camfield P, Camfield C. A schematic approach to hypotonia in infancy.Paediatr Child Health. 2005; 10(7):397–400
10. Visser VE, Hall RT. Urine culture in the evaluation of suspected neonatal sepsis.J Pediatr. 1979;94(4):635–638
11. Engorn B, Flerlage J, eds.The Harriet Lane Handbook E-Book. 20th ed. Philadelphia, PA: Elsevier Health Sciences; 2014
12. Bodensteiner JB. The evaluation of the hypotonic infant.Semin Pediatr Neurol. 2008;15(1):10–20
13. Peredo DE, Hannibal MC. Thefloppy infant: evaluation of hypotonia.Pediatr Rev. 2009;30(9):e66–e76
14. Prasad AN, Prasad C. Genetic evaluation of thefloppy infant.Semin Fetal Neonatal Med. 2011;16(2):99–108
15. Saudubray JM, Nassogne MC, de Lonlay P, Touati G. Clinical approach to inherited metabolic disorders in neonates: an overview.Semin Neonatol. 2002;7(1):3–15
assignment of the gene and identification in four patients of nine different mutations within the VLCAD gene.Hum Mol Genet. 1996;5(4): 461–472
17. Leslie ND, Valencia CA, Strauss AW, Zhang K, et al. Very long-chain acyl-coenzyme A dehydrogenase deficiency. In: Adam MP, Ardinger HH, Pagon RA, eds.GeneReviews. Seattle, WA: University of Washington; 2009
18. Andresen BS, Olpin S, Poorthuis BJ, et al. Clear correlation of genotype with disease phenotype in very-long-chain
acyl-CoA dehydrogenase deficiency.Am J Hum Genet. 1999;64(2):479–494
19. Solis JO, Singh RH. Management of fatty acid oxidation disorders: a survey of current treatment strategies.J Am Diet Assoc. 2002;102(12):1800–1803
20. Leonard JV, Morris AA. Diagnosis and early management of inborn errors of metabolism presenting around the time of birth.Acta Paediatr. 2006;95(1):6–14
21. New England Consortium of Metabolic Programs. Acute illness protocols. Available at: https://
newenglandconsortium.org/for-professionals/acute-illness-protocols/. Accessed May 13, 2019
22. Adam MP, Ardinger HH, Pagon RA, et alet al, eds.GeneReviews. Seattle, WA: University of Washington;
2010
23. IEMbase. Inborn errors of metabolism knowledgebase. Available at: http:// iembase.org/. Accessed July 30, 2018
24. American College of Medical Genetics.
DOI: 10.1542/peds.2018-0788 originally published online June 21, 2019;
2019;144;
Pediatrics
and Jodi D. Hoffman
Adrienne H. Long, Jennifer G. Fiore, Riaz Gillani, Laurie M. Douglass, Alan M. Fujii
Hypotonia and Lethargy in a Two-Day-Old Male Infant
Services
Updated Information &
http://pediatrics.aappublications.org/content/144/1/e20180788
including high resolution figures, can be found at:
References
http://pediatrics.aappublications.org/content/144/1/e20180788#BIBL
This article cites 18 articles, 6 of which you can access for free at:
Subspecialty Collections
http://www.aappublications.org/cgi/collection/neonatology_sub
Neonatology
sub
http://www.aappublications.org/cgi/collection/fetus:newborn_infant_
Fetus/Newborn Infant following collection(s):
This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtml
in its entirety can be found online at:
Information about reproducing this article in parts (figures, tables) or
Reprints
http://www.aappublications.org/site/misc/reprints.xhtml
DOI: 10.1542/peds.2018-0788 originally published online June 21, 2019;
2019;144;
Pediatrics
and Jodi D. Hoffman
Adrienne H. Long, Jennifer G. Fiore, Riaz Gillani, Laurie M. Douglass, Alan M. Fujii
Hypotonia and Lethargy in a Two-Day-Old Male Infant
http://pediatrics.aappublications.org/content/144/1/e20180788
located on the World Wide Web at:
The online version of this article, along with updated information and services, is
by the American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.