C o m p u tation al A n alysis o f
P ro tein S equence and Stru ctu re
R ob ert M a tth ew M acC allum
September 1997
Biomolecular Structure and Modelling Group
Department of Biochemistry and Molecular Biology
University College London
Gower Street
London W CIE 6BT
ProQuest Number: 10055396
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest 10055396
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author.
All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
A b stract
This project has combined the structural and functional analysis of proteins, protein structure prediction, and sequence analysis.
A study of antibody-antigen interactions has been undertaken. We have anal ysed antigen-contacting residues and combining site shape in the antibody crystal structures available in the Protein D ata Bank. Antigen-contacting propensities are presented for each antibody residue, allowing a new definition for the complemen tarity determining regions to be proposed based on observed antigen contacts. An objective means of classifying protein surfaces by gross topography has been devel oped and applied to the antibody combining site surfaces. The surfaces have been clustered into four topographic classes, confirming suggestions in the literature th at antigen type might influence the shape of the whole combining site.
The prediction of secondary structural class and architecture from sequence composition analysis has also been investigated. Modifications to a well estab lished geometric prediction algorithm have led to improvements in accuracy and the estimation of reliability. The hierarchical prediction of fold architectures using these methods is then discussed.
To complement the ab initio approach of class and architecture prediction, a novel sequence alignment algorithm employing direct comparisons of predicted sec ondary structure and sequence-derived hydrophobicity was developed, and applied to fold recognition. The method, called SIVA, appears to perform well when suffi cient multiple sequence information is available, although further testing, including blind public testing, is required.
A b b reviation s
3D,2D,ID
CASP
CATH
CDR
DNA
DSC
DSSP
GA
GOR
NMR
PDB
PHD
RMS
RNA
SCOP
SIVA
SSAP
SSCP
SWISS-PROT
TIM
Three-dimensional, etc.
Critical Assessment of Structure Prediction (meeting)
Structure classification (Orengo et al., 1997)
Complementarity determining region
Deoxyribonucleic acid
Secondary structure prediction algorithm (King & Sternberg, 1996)
Dictionary of Protein Secondary Structure (Kabsch & Sander, 1983)
Genetic algorithm
Gamier Osguthorpe Robson (secondary structure prediction method)
Nuclear magnetic resonance
Protein Data Bank
Secondary structure prediction algorithm (Rost &: Sander, 1994)
Root mean square
Ribonucleic acid
Structural Classification of Proteins (Murzin et al., 1995)
Sequence-derived Information Vector Alignment
Structural alignment algorithm (Taylor & Orengo, 1989)
Secondary Structural Content of Proteins (Eisenhaber et al., 1996a)
Sequence database (Bairoch & Boeckmann, 1991)
A ck n ow led gem ents
C ontents
In trod u ction 13
1.1 Protein structure and fu n c tio n ... 14
1.1.1 Prim ary s tr u c tu r e ... 14
1.1.2 Secondary s tru c tu re ... 15
1.1.3 Tertiary s tr u c tu r e ... 17
1.1.4 Q uaternary structure — macromolecular assem b lies... 18
1.1.5 Soluble, fibrous and membrane p ro te in s ... 19
1.1.6 E n z y m e s... 19
1.1.7 Electron transfer proteins ... 20
1.1.8 Proteins in signalling and re c o g n itio n ... 20
1.1.9 Mechanical p ro te in s ... 21
1.1.10 Regulation and control of protein f u n c t i o n ... 21
1.2 Protein structure classification... 22
1.2.1 The dynamic programming alignment a lg o rith m ... 23
1.2.2 Structural a lig n m e n t... 25
1.2.3 Classification schemes ... 26
1.3 Molecular interactions of p r o te in s ... 29
1.3.1 Protein surfaces and b in d in g ... 29
1.3.2 Molecular surfaces ... 31
1.3.3 D o c k in g ... 31
1.4 Prediction of protein structure and f u n c t i o n ... 32
1.4.1 Sequence methods ... 32
1.4.2 Fold reco g n itio n ... 35
1.4.3 Comparative m o d ellin g ... 38
1.4.4 Ab initio m e th o d s... 38
1.4.5 CASP - Critical Assessment of Structure P re d ic tio n 45 1.5 Outline of th e s is ... 46
A n tib o d y -a n tig en in teraction s 47 2.1 In tro d u c tio n ... 47
2.2 Methods and D a t a ... 48
2.2.1 Antibody structures and s e q u e n c e s ... 48
Contents 6
2.2.3 Canonical loops and structural alig n m e n t... 50
2.2.4 Surface a n a ly s is ... 50
2.3 R e s u lts ... 51
2.3.1 Contact A n a ly s is ... 51
2.3.2 Antibody surface topography ... 59
2.3.3 Predicting antigen contacts ... 64
2.4 D iscu ssio n ... 67
3 C lass and a rch itectu re p red ictio n 70 3.1 In tro d u c tio n ... 70
3.2 Class p re d ic tio n ... 71
3.2.1 Overview of a l g o r it h m ... 71
3.2.2 D ataset s i z e ... 72
3.2.3 D ataset completeness and class d e fin itio n s ... 72
3.2.4 Pairwise s i m i l a r i t y ... 74
3.2.5 Jac k -k n ifin g ... 74
3.2.6 Sequence length and n o i s e ... 75
3.2.7 Multiple s e q u e n c e s ... 77
3.2.8 N-tuple c o m p o s itio n ... 78
3.2.9 Prediction confidence... 79
3.2.10 Helix/non-helix p r e d ic tio n ... 80
3.3 Secondary structural content p re d ic tio n ... 81
3.4 Hierarchical class and architecture p re d ictio n ... 82
3.4.1 Distinction between m ainly-a and mixed-a/? c la s s e s ... 83
3.4.2 Prediction of m ainly-^ arch itectu res... 84
3.4.3 Prediction of other arc h ite ctu re s... 87
3.5 Origins of sequence composition differences... 87
3.5.1 Methods — Analysis of sequence pattern usage ... 87
3.5.2 Secondary structural c l a s s ... 90
3.5.3 Mainly-/) a rc h ite c tu re s ... 94
3.6 D iscu ssio n ... 97
4 A lig n m en ts o f m u ltip le seq uence-d erived p rop erty v ecto rs 100 4.1 In tro d u c tio n ... 100
4.1.1 Sequence properties and derived in fo rm a tio n ... 102
4.2 Methods and D a t a ... 105
4.2.1 Fold library and query sequences ...105
4.2.2 Evaluation of fold recognition p e rfo rm a n c e ...108
4.2.3 Evaluation of alignment q u a l i t y ... 109
4.2.4 Sequence alignment a lg o r ith m ... 109
Contents 7
4.3.3 Sequence C o n se rv atio n ... 114
4.3.4 Secondary structure p r e d ic tio n s ...118
4.4 Analysis and D is c u s s io n ... 121
4.4.1 Respectable fold recognition from sequence a lo n e ? ... 121
4.4.2 Length dependence and local a lig n m e n ts ... 125
4.4.3 The Fischer and Eisenberg b e n c h m a r k ...126
4.4.4 Significance estimates and null p re d ic tio n s ... 128
4.4.5 Alignment q u a lity ...132
4.4.6 Future developments of S I V A ... 134
5 Self-organising m aps o f p rotein stru ctu re and sequence 136 5.1 In tro d u c tio n ... 136
5.2 Self-organising maps - Overview and Methods ... 137
5.2.1 Im p le m e n ta tio n ... 138
5.2.2 Visualisation ... 139
5.3 Mapping of three-dimensional protein s t r u c t u r e ... 140
5.3.1 Self-organising maps of two superfolds ... 140
5.3.2 Map disco n tin u ities...142
5.3.3 Map dim ensions... 143
5.3.4 Fold e sse n c e ...145
5.4 Mapping of structure and sequence p r o p e r t i e s ...147
5.4.1 Sequence-derived hydropathy ... 147
5.4.2 Sequence c o n s e rv a tio n ... 149
5.4.3 Predicted secondary structure in fo r m a tio n ... 153
5.5 Alignment of structure-m apped sequence in fo rm a tio n ...155
5.5.1 Methods — fold recognition using mapped sequences . . . . 155
5.5.2 Results — fold recognition using mapped s e q u e n c e s... 156
5.5.3 Alignment quality for a/3 doubly wound folds ...158
5.5.4 Gap p e n a ltie s ... 160
5.6 Analysis and D is c u s s io n ...161
5.6.1 Possible d e v e lo p m e n ts ... 162
6 C on clu d in g rem arks 166 A G ath erin g m u ltip le sequences for p ro tein dom ains 169 A .l O verview ... 169
A.2 Domain sequences...170
A.3 Database s e a rc h ... 170
A.4 Multiple sequence a l ig n m e n ts ... 171
A.5 Indel r e m o v a l... 171
Contents 8
B C A T H description s 173
List o f Figures
1.1 The dynamic programming a lg o rith m ... 24
1.2 Definitions of molecular su rfa c e s... 32
2.1 Analysis of light chain antigen c o n ta c ts ... 54
2.2 Analysis of heavy chain antigen c o n t a c t s ... 55
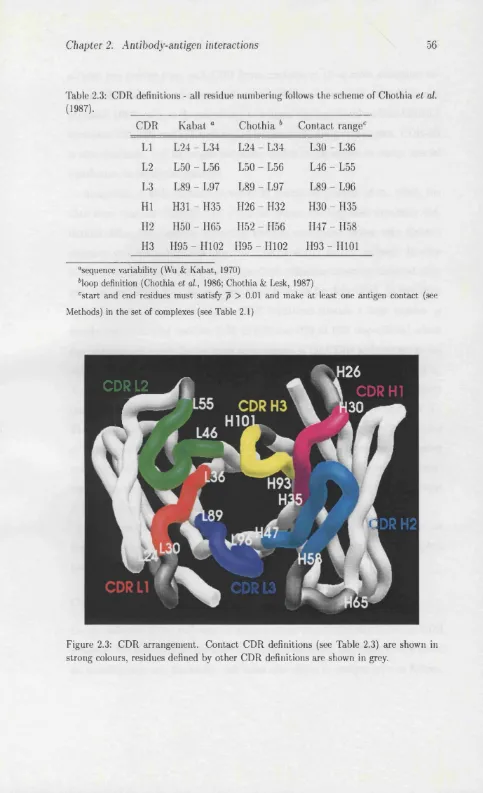
2.3 CDR arrangement ... 56
2.4 Antigen contacts and CDR c o n fo rm a tio n ... 59
2.5 Antigen contacts compared between different loop conformations . . 60
2.6 Combining site shape a n a ly s is ... 61
2.7 Antigen binding pocket p re d ic tio n ... 66
3.1 Reducing noise in the prediction d a ta s e ts ... 76
3.2 Reliability measures for class p r e d ic tio n s ... 80
3.3 Common mainly-/? a rc h ite c tu re s ... 85
3.4 Location of significant sequence patterns in protein structures . . . 93
4.1 Fold recognition performance using hydrophobicity and conservation 120 4.2 Fold recognition performance using hydrophobicity and DSC sec ondary structure p re d ic tio n s ...123
4.3 Summary statistics of fold recognition t r i a l s ... 129
4.4 Plots of fold recognition results vs. structural similarity measures . 133 5.1 Kohonen map overview ... 138
5.2 Mapping of domain lcgt04 three-dimensional c o o rd in a te s ... 141
5.3 Mapping of domain IghsAO three-dimensional c o o r d i n a t e s ...141
5.4 Distance m atrix from self-organising m a p ...143
5.5 Mapping discontinuities ...144
5.6 Protein shape and map d im e n s io n s ... 146
5.7 Sequence derived hydropathy for Is x a A O ...148
5.8 Structure/H ydropathy mapping ( i ) ... 150
5.9 Structure/H ydropathy mapping ( i i ) ... 151
5.10 Hydropathy mapping of a selection of d o m a in s ...152
5.11 Mapping sequence conservation and structure ...154
List of Figures 10
List o f Tables
2.1 Complexed antibody s t r u c t u r e s ... 52 2.2 Uncomplexed antibody s tr u c tu r e s ... 53 2.3 CDR d e fin itio n s... 56 2.4 Combining site shape classes for the complexed antibody structures 63 2.5 Combining site shape classes for the uncomplexed antibody structures 63 2.6 Shape classes of complexed and uncomplexed a n t i b o d i e s ... 65 3.1 Secondary structural class prediction a c c u r a c ie s ... 73 3.2 Secondary Structural Content Prediction using a modified version
of SSCP (Eisenhaber et a i , 1996a; Eisenhaber et a i , 1996b) . . . . 82 3.3 Mainly-/^ architecture p red ictio n ... 86 3.4 Xt analysis of amino acid usage between the three main classes . . . 91 3.5 X? analysis of (i,i+3) duplet usage between the three main classes . 92 3.6 Xt analysis of (i,i+3) duplet usage between the mainly alpha and
mixed alpha-beta c la s s e s ... 95 3.7 Xt analysis of (i,i+3) duplet usage between the ribbon and non
ribbon beta a rc h ite ctu re s... 95 3.8 Xt analysis of (i,i+3) duplet usage between the sandwich, barrel and
distorted sandwich a rc h ite c tu re s... 96 4.1 Fold recognition library/query d o m a in s ...106 4.2 Correct top-ranking fold recognition results using Smith W aterman
local a lig n m e n ts... 113 4.3 Four hydrophobicity related sc a le s ... 114 4.4 Summary of fold recognition using alignment of raw sequence de
rived inform ation...115 4.5 Summary of fold recognition results using alignments of hydropho
bicity (Kyte and D o o little )...116 4.6 Summary of fold recognition results using alignments of hydropho
bicity (Bull and Breese) ... 117 4.7 Summary of fold recognition results using alignments of hydrophobic
moment d ir e c tio n ... 118 4.8 Summary of fold recognition results using alignments of conserved
hydrophobicity s c o r e ...119
List of Tables 12
4.9 Summary of fold recognition results using alignments of combined raw hydrophobicity and DSC secondary structure prediction prob
abilities ... 122
4.10 Similar folds with different CATH topologies are recognised...130
B .l CATH architectures...174
B.2 Descriptions for selected m ainly-a CATH to p o lo g ies... 175
B.3 Descriptions for selected mainly-^ CATH to p o lo g ie s... 175
C hapter 1
In trod u ction
Living organisms are the product of a complex interplay of cellular processes. These processes are controlled through the interactions of molecules within and between cells. Much progress has been made in understanding the details of these phe nomena through basic research in many areas of experimental biology. Yet our understanding of biological systems as a whole is largely superficial. The parallel, interconnected and apparently redundant nature of many processes renders them intractable to dissection by standard techniques. The field known as ‘integrative biology’ has been born out of the need to assimilate information from diverse fields and formulate more complete theories to explain the living world.
Com putational biology will have a central role in this field because of the ease with which computers can handle large amounts of data and probe the non-linear dynamics of nature. The experimental determ ination of macromolecular structures has helped to unravel the mysteries of many cellular processes. Three dimensional structure, intermolecular interactions and function are closely linked. Unfortu nately, the networks of interactions between molecules in vivo do not always fit the simple models we are often forced to assume. Computer simulation of complex systems like these is possibly the only route towards determining their behaviour. This approach will require a deeper understanding of interactions between biolog ical molecules, and faster routes to the determ ination of their structures.
The m ajority of the complex cellular machinery is protein. O ther components, such as nucleic acid, carbohydrate and lipid, also play im portant roles, however
Chapter 1. Introduction 14
proteins perform the widest array of functions throughout the cell and beyond. Genome sequencing projects around the world (Fleischmann et al., 1995; Fraser et al, 1995; Bult et al, 1996, for example) are rapidly collecting genetic information from a number of organisms. The genome completely specifies the construction and maintenance of an organism primarily through the encoding of its proteins, collectively known as the proteome. Consequently, biochemists are faced with the task of translating genome and proteome into a functional description of cells and organisms. The work described in this thesis is part of this effort to break the code.
1.1
P rotein structure and function
Proteins are made in the cells of plants, animals and bacteria by a universal mech anism. The DNA* of an organism encodes its proteins in a linear sequence of nucleotides of which there are four types: adenine, cytosine, guanine and thymine. This information is copied to an intermediate nucleic acid form (messenger RNA), which is then linearly processed during protein synthesis. Nucleic acid and protein synthesis typify the complexity of the cellular machinery; hundreds of proteins are involved. This section provides an overview of protein structure and its relationship to function.
1.1.1
P rim ary structure
As the messenger RNA molecule is linearly processed by the protein synthesis machinery (the ribosome), each non-overlapping triplet of nucleotides, known as a codon, specifies a corresponding subunit, or residue, to be added to the growing polypeptide chain. Each residue can be any one of the 20 standard amino-acids; the correspondence between the sequence of nucleotides of the codon and the amino acids is constant in all but a few organisms and is known as the genetic code. W ith the exception of proline (which is actually an imino acid) the amino acids share the common feature of an amino and carboxyl group joined by a single carbon atom.
Chapter 1. Introduction 15
known as the carbon-a atom, from which different side-chains are attached (the amino acid glycine has no side-chain).
The polypeptide chain is a linear heteropolymer of these amino acids linked by the covalent peptide bond between the carboxyl carbon atom and the nitrogen of the amino group. The peptide bond does not rotate freely, but the other two backbone bonds can rotate, allowing the polypeptide chain to fold in almost any direction. Proteins generally have more than about 20 residues; shorter chains are called peptides. The different physical and chemical properties of the side-chains determine both the local and global conformations adopted by polypeptide chains discussed below.
1.1.2
Secondary stru cture
W hilst peptides do not always exhibit ordered three-dimensional structure, most proteins consistently fold into the same configuration. The three-dimensional struc ture of proteins is uniquely determined by its prim ary structure (Anfinsen, 1973), although the folding process can be aided by molecules known as chaperones (Hartl, 1996). The first experimentally determined protein structure, the X-ray diffraction structure of myoglobin by Kendrew (1958), showed the polypeptide chain bundled into a compact tertiary structure. For an introduction to protein structure de termination, see Chapter 17 in Branden and Tooze (1991). Subsequent higher resolution structures of myoglobin (Kendrew et ai, 1960) confirmed the existence of a regular helical arrangement, called a-helix, in much of the polypeptide chain, proposed by Pauling and Corey (Pauling et ai, 1951) from theoretical studies of peptides and X-ray analysis of hair fibres. At th a t time, Pauling and Corey (1951) had also correctly predicted the existence of sheet-like structures of non-covalently cross-linked strands of extended polypeptide chain which they called /3-sheet,
Chapter 1. Introduction 16
a -h e lic e s
As the polypeptide chain coils in a right handed manner, the CO and NH groups of residues i and 2 + 4 respectively form hydrogen bonds which stabilise the helix. All or most of residues in a helix are bonded in this way, making it a relatively rigid unit of structure with very little free space in its core. The helix makes a whole tu rn per 3.6 residues, and can have between 4 and around 50 residues.
/^-sheets
The most regular form of extended polypeptide chain seen in protein structures is the /^-strand, /^-strands are stabilised though interactions with other ^-strands to form a j3-sheet, which is more strictly a tertiary structure. Like helices, sheets too are stabilised by hydrogen bonds between CO and NH groups, but in this case they are much more distantly separated along the chain. Both parallel and anti-parallel /^-sheets exist in protein structures. Due to the geometry of the peptide backbone, the amino acid side chains of /^-strands alternate on either side of the sheet.
T u rn s
The polypeptide chain can make very sharp changes in direction using as few as four residues by means of hydrogen bond stabilised /^-turns. These secondary structures often have specific amino acid preferences (Hutchinson & Thornton, 1994) and commonly contain proline an d /o r glycine residues.
O th e r s e c o n d a ry s tr u c tu r e s
Chapter 1. Introduction 17
1.1.3
Tertiary structure
The process of protein folding results in a compact structure in which secondary structure elements are packed against each other in a stable configuration, often called a Told’. The tendency for the burial of hydrophobic side-chains in the inte rior, or core, of proteins has been observed in nearly all structures solved to date, and is believed to be the driving force of tertiary structure formation (Dill, 1990). Other interactions help to stabilise the fold: hydrogen bonds, van der Waals forces, and oppositely charged amino acid side-chains forming salt-bridges. Many elegant structures have evolved, including curved, barrel-like /^-sheets, parallel bundles of helices, and propellor-like structures. Folds are also referred to as Topologies’ since they can be thought of as sets of connected secondary structure elements.
D om ains
Many longer polypeptide chains appear to fold into more than one distinct unit, or domain. Domains can only be defined subjectively, but they are usually unambigu ously distinguishable by eye as self-contained units of structure, and have distinct hydrophobic cores. The autom atic definition of domains (Swindells, 1995b; Islam
et al., 1995; Siddiqui & Barton, 1995) is currently unreliable, despite the obvious
need for such methods as the number of determined protein structures continues to grow.
D isu lp h id e bonds
Chapter 1. Introduction 18
S e c o n d a ry s tr u c t u r a l class
In 1976 Levitt and Chothia observed th a t the protein structures then available seemed to group naturally into four classes based upon the gross secondary struc tural content of their tertiary structures. These classes were: m ainly-a, mainly-/?, alternating a / (3 and a + (3 (separated along chain, not alternating). W hilst being a largely subjective definition, it has served well in the intervening years.
Recently, in conjunction with the construction of a classified database of do mains (see Section 1.2), an autom ated approach to this deceptively difficult prob lem was developed (Michie et ai, 1996). It was found th a t the distributions of the two mixed-a/? classes were continuous and overlapping using any of a number of structure-derived parameters. Furthermore, the database of domains had reduced the number of a j3 examples through their division into separate m ainly-a and mainly-/? domains. Thus when dealing with domains it is acceptable to use a three class definition: m ainly-a, m ainly-^ and mixed-a/?. Some proteins contain little or no regular secondary structure, these are often termed ‘irregular’ proteins.
S u p e r-s e c o n d a ry m o tifs
As the number of solved structures began to increase it became apparent th a t different folds often possessed similar arrangements of a two to four consecutive secondary structures. These recurring units are called super-secondary structures (Rao & G., 1973) or motifs. Possibly the most common super-secondary motif is the
j3aj3 unit (Sternberg & Thornton, 1976), found in folds with parallel /^-sheets. The
/?-hairpin (two /?-strands connected by a turn) and the four-strand “Greek key” motif (Richardson, 1981; Richardson, 1986) are other common super-secondary structures.
1.1.4
Q uaternary structure — m acrom olecular assem blies
Chapter 1. Introduction 19
of oxygen transport to the tissues. Haemoglobin contains four non-protein haem prosthetic groups which are non-covalently bound and actually bind oxygen. The subunits of haemoglobin are of similar sequence and structure, more heterogeneous complexes are also common (RNA polymerases, viral coats, electron transfer complexes). The importance of interactions such as these in protein function is discussed in more detail in Section 1.3.
1.1.5
Soluble, fibrous and m em brane protein s
In the field of structural biology it is easy to forget th a t the m ajority of protein structures determined to date help to explain only part of cellular protein function. The determ ination of protein structures to atomic resolution currently requires pro teins to be water soluble, and to either crystallise (for X-ray crystallography) or be smaller than around 200 residues (for NMR spectroscopy). Many im portant structural proteins in the extracellular m atrix are insoluble: collagen, elastin and keratin, for example. These are classed as fibrous proteins. The phospholipid cell, endoplasmic and nuclear membranes are foci for the m ajority of cellular events. The membrane provides a medium for the co-localisation of proteins, some of which span the membrane one or more times. However, the folding, structure and sta bility of membrane spanning proteins are generally dependent on the membrane environment and this makes them difficult to solubilise intact for structure determi nation. A handful of membrane proteins have been crystallised however, including the photosynthetic reaction centre of Rhodopseudomonas viridis (Deisenhofer et ai, 1985), a purple sulphur bacterium, and porin, the outer membrane channel protein of mitochondria (Weiss & Schulz, 1992).
1.1.6
E nzym es
Chapter 1. Introduction 20
from the modification of small organic molecules such as in the synthesis of amino acids, to the degradation of large proteins. Many enzymatic reactions require the exclusion of water, and so the catalytic surface, or active site, is often recessed in a cleft formed from hydrophobic side chains. Active site residues are usually well conserved in the sequences of related proteins with similar functions. Interestingly, some possibly unrelated proteins with similar reaction mechanisms show spatial conservation in three dimensional relationships of active site residues (Wallace et
ai, 1996).
1.1.7
E lectron transfer proteins
Photosynthesis and respiration are examples of mechanisms in the cell which in volve the transfer of energy through space from one macromolecular component to another in the form of electrons. W hilst the arom atic side amino acid side chains of proteins have delocalised electron fields and can accept and transfer ex cess charge, most electron transfer groups utilise prosthetic groups such as haem and iron-sulphur clusters, which contain many more atoms with delocalised elec trons. Delocalisation and electron transfer also requires the exclusion of water, and this is provided by the surrounding protein structure.
1.1.8
P ro tein s in signalling and recognition
Chapter 1. Introduction 21
1.1.9
M echanical proteins
In addition to roles as catalysts and signalling molecules, proteins also perform the more modest role as structural or mechanical proteins. The fibrous extracel lular m atrix proteins have already been mentioned, but inside the cell there are many more. The histones are small globular proteins around which chromosomal DNA wraps itself for efficient packaging during nuclear division. DNA packaging probably also has a role in gene expression. The uptake of molecules into cells by endocytosis involves the invagination of the cell membrane to form vesicles. Vesicle formation throughout the cell is aided by proteins like clathrin which associate in a polyhedral cage-like structure and promote the curvature of the membrane.
Force generating cellular machinery also exists: muscle is a linear m otor which utilises conformational change and a ratchet-like mechanism. Intracellular trans port along cytoskeletal microtubules uses a similar mechanism. Breathtakingly elegant rotary mechanisms also exist. The bacterial fiagellum is a large multi protein assembly which can rotate at over 100 revolutions per second. The recently solved structure of the F I subunit of ATP synthase (Abrahams et al., 1994; Noji
et al., 1997) showed th a t rotary mechanisms are possible in much smaller protein
assemblies, in this case involving only 7 polypeptide subunits.
1.1.10
R egu lation and control o f protein function
Chapter 1. Introduction 22
Steric effects
In some cases, enzymes will have a binding site for a regulatory protein in the vicinity of the active site. When the regulator binds, it blocks access to the active site for the substrate. This is called competitive inhibition. The same type of inhibition can prevent two binding partners from associating to form an active multimeric protein. Conversely, the association of subunits can enhance function. Protein modifications, such as phosphorylation, can have similar steric effects.
C onform ational changes
Regulator binding and protein modification far from the functional region of a protein can also modulate its function, through conformational dynamics. As the regulator binds, structural changes are propagated through the molecule, altering the conformation of functional residues. In these proteins, there exist more than one stable conformation in dynamic equilibrium, and the m odulatory signal tips the balance between those states. The cooperativity of oxygen binding to haemoglobin discussed earlier is the result of conformational changes transm itted from one haem group to another through the protein as a whole.
1.2
P rotein stru cture classificatiou
Chapter 1. Introduction 23
1.2.1
T h e dynam ic program m ing alignm ent algorithm
The first application of dynamic programming to biological sequence alignment (both DNA and protein) was by Needleman and Wunsch (1970). This and re lated algorithms have been in use since then for the detection of similarities (see Section 1.4.1) and the alignment of sequence information from protein families.
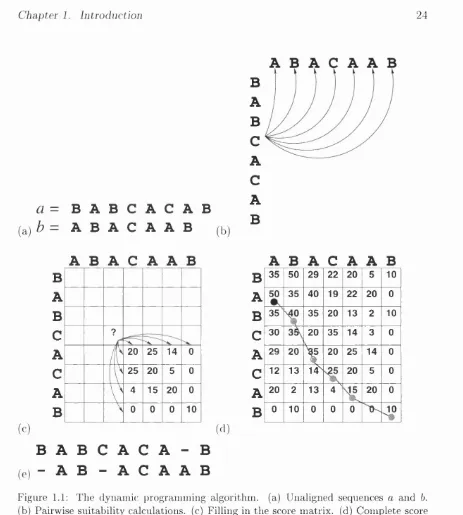
Figure 1.1(a) shows two short (imaginary) sequences we wish to align: sequence
a = BABCACAB, sequence b = BCCAAB. Residues i and j in sequences a and
b are denoted by and bj respectively. Similarly their lengths are denoted m and n. The objective of any alignment algorithm is to maximise the alignment of similar or identical residues in a with b, equivalent to X) S ij along the path of the alignment. S i j is best explained as how ‘happy’ residue is aligned against bj.
In sequence comparison, the value of S i j is looked up in a m atrix M containing the log-likelihoods of all substitutions between each of the 20 amino acids (20 x 20 = 400). M is approximated from the frequencies of substitutions observed in sequence alignments (Barker & Dayhoff, 1972; Dayhoff et ai, 1978; Henikoff & Henikoff, 1992). Thus S ij = Ma^^bj and when = bj, Ma^^bj gives a high positive value. Smaller or negative values of Ma^^bj occur when the amino acids and bj are less alike in terms of observed substitutions.
Gapless alignment of sequence a and b would simply involve sliding one sequence along the other and outputting the alignment where S ij is maximal, involving a number calculations only linearly dependent upon the sequence lengths. The allowance of gaps requires th at each 0% must be ‘tested’ against bj. Figure 1.1(b) illustrates the number of possibilities which exist in the alignment of residue (C) with sequence b. There are m x n such comparisons in total. Thus gapped alignments involve calculations scaling approximately to the square of number of residues.
Chapter 1. Introduction 24
(a)
a
=B A B C A C A B
b = A B A C A A B
(b)
B
A
B
C
A
C
A
B
A B A
cA A B
A B A
cA A B
B
B
35 50 29 22 20 5 10A
A
50 35 40 19 22 20 0B
B
35 35 20 13 2 10C
?C
30 20V
35 14 3 0
A
i
20 25 14 0A
29 20X
20 25 14 0C
25 20 5 0C
12 13 14^ 20 5 0A
V
4 15 20 0A
20 2 13 4 20 0B
\ 0 0 0 10B
0 10 0 0 04»
( d ) (c)
B A B C A C A - B
( e) “ A B - A C A A B
Figure 1.1: The dynamic programming algorithm, (a) Unaligned sequences a and b.
(b) Pairwise suitability calculations, (c) Filling in the score matrix, (d) Complete score
matrix and alignment trace-back, (e) Alignments between a and b. Scoring with identity
matrix (identity=10, otherwise=0), gap open penalty = 5 and gap extension penalty = 1. See text for full explanation.
starting from the bottom right-hand corner as follows:
m a X p ^ 2 — (ffo + f f e i p — i - 2 ) ) ) ,
max,^^2 - (Qo + 9e[(l — j - 2)))
Chapter 1. Introduction 25
where Qq and Çe are fixed penalties for the creation and extension of gaps, respec tively. Figure 1.1(c) shows the m atrix D in the example partially filled in, and the scores from which Dg 2 are inherited. In this example the M is the simple identity m atrix (scoring 10 for identity and 0 otherwise); Qo = and = 1.
When complete, the highest value from the top and left edges of the m atrix D
is found (marked with a black filled circle in Figure 1.1(d)) and the path which resulted in this score is traced back in reverse to generate the alignment, shown in Figure 1.1(e). A relatively minor modification to this algorithm was developed by Smith and W aterman (Smith & W aterman, 1981) to locate the best local align ments between two sequences. Local alignments are not required to include the termini of either sequence, as in the Needleman and Wunsch (1970) m ethod which is a global alignment.
1.2.2
Structural alignm ent
Structural alignment is also possible using the basic dynamic programming algo rithm . The value of S ij could be calculated from the RMS deviation of fixed windows centred around Ai and Bj fitted by least-squares superposition of atomic coordinates. Taylor and Orengo (1989) found, however th a t this approach would not take into account long range interactions with small windows and larger win dows lacked the fiexibility to accommodate insertions and deletions. Their solution, in the program SSAP, was to apply the dynamic programming algorithm to the scoring of similarity between the structural environments around Ai and Bj. Sij
is defined as the total score of the low level alignment whose equivalent pairwise suitability measure Lk^i is calculated from the distance between interatom ic vectors
Vik and Vji defined using a common reference frame around carbon-as.
Chapter 1. Introduction 26
ming algorithm to generate the minimum soap-film area (Schulz, 1977) between arbitrarily superposed carbon-a backbones. The soap film area is then minimised through the sampling of superposition parameters, producing alignment and su perposition simultaneously.
The DALI program by Holm and Sander (Holm & Sander, 1993) uses simulated annealing to generate alignments of structural fragments, and has the interesting feature of being able to find alignments involving chain reversals and different topologies.
1.2.3
C lassification schem es
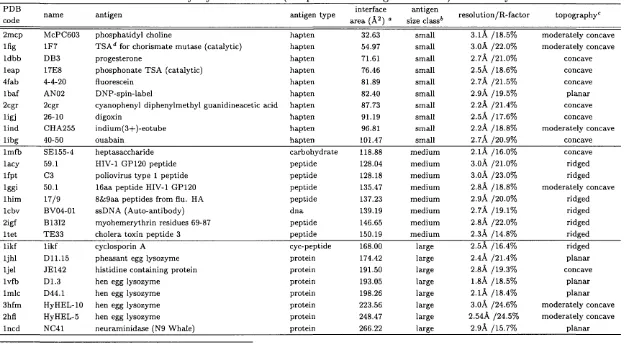
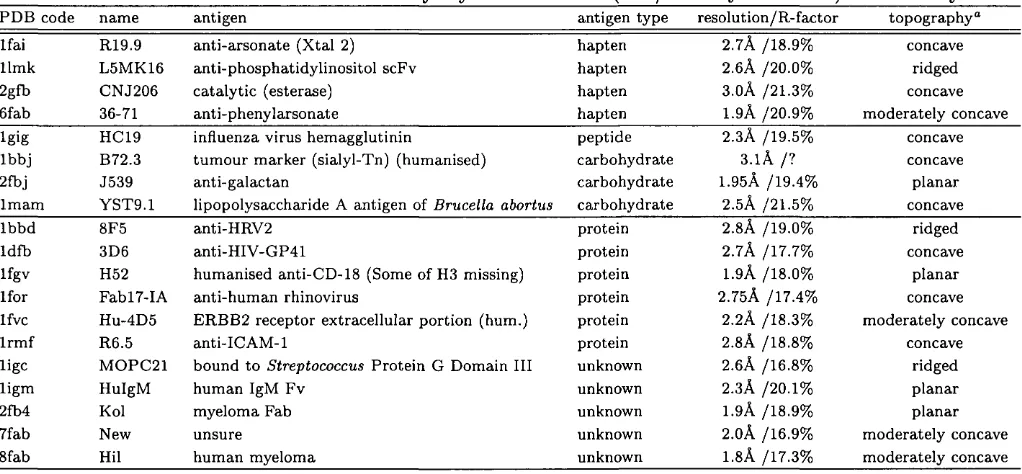
Many of the laboratories which have developed structure comparison algorithms have also undertaken the difficult task of classifying protein structures. The Pro tein D ata Bank, or PDB (Bernstein et al., 1977), is the most obvious source of material, since it is where the m ajority of experimentally determined structures are deposited and is freely available to all*. There are currently in the order of 5000 crystallographic protein structures and 900 NMR protein structures in the PDB; added to which are a number of nucleic acid, carbohydrate and peptide structures and theoretical models. Pre-processing and pairwise comparison of the proteins in the PDB is a major task. As discussed above, the splitting of multi domain proteins into their constituent domains is difficult to autom ate, yet it is an essential part of any classification effort.
SC O P
As a result, one of the most accurate classifications of protein structures, the SCOP database (Murzin et al, 1995), is largely constructed by hand. Expert knowledge of protein structure and biochemistry is translated into an internet resource^ which provides information about folds and their probable evolutionary relationships. It is also kept remarkably up to date, usually being no more than a few months behind the PDB. Since information is sourced from the literature, the database may even
*http://www2.ebi.ac.uk/pdb/
Chapter 1. Introduction 27
describe folds whose coordinates are not yet released to the public.
Autom ated classifications offer much faster production times and more effi cient use of resources (computer time is cheap). They fail however where manual methods succeed, in the classification of proteins with similar folds, but different functions. Numerical cutoffs and alignment param eters are no match for expert knowledge and the human brain. Nonetheless, two very useful structure databases exist which are best described as ‘sem i-autom ated’.
F S S P
The FSSP database of the Sander group (Holm & Sander, 1994) uses the DALI structure comparison program to define a non-redundant set of protein structures from the PDB. It does not currently consider domains as separate entities. This database can be searched with a newly solved structure as the query, allowing crystallographers and NMR spectroscopists to draw parallels to the structures and functions of known proteins of which they are unaware.
C A T H
The CATH* database from this laboratory (Orengo et ai, 1997) is a more ambitious project. Using semi-automated methods a set of domains from the single- and multi-domain structures of the PDB is hierarchically classified at four levels: Class, Architecture, Topology and Homologous superfamily. These levels are described both by names (for ease of human understanding) and numbers (for easy computer manipulation). In this latter form CATH is similar to the EC enzyme classification scheme (NC-IUBMB, 1992).
Class has already been described and can be one of: m ainly-a, mainly-/?, mixed-
a(5 and irregular. The unique feature of CATH is the architectural description at the next level in the hierarchy. Many of the folds (in the mainly-/? and mixed-a/? classes in particular) appear to be constructed according to the same basic princi ples. For example a sizeable subset of mixed-a,(? folds can be thought of as having three layers: two of helix surrounding a single central layer of ^-sheet; this is the
Chapter 1. Introduction 28
layer a/3a sandwich architecture. Architectures are defined manually for the whole of fold space. Folds within the same architectural subdivision may have different numbers and ordered connections of secondary structures, and are discriminated by the topology descriptor. The final level groups all domains belonging to the same homologous superfamily. These are structures which are clearly or weakly related by sequence but have the same function and are most likely evolutionarily related.
The topology and homology classifications of CATH are performed by the auto m ated application of the SSAP algorithm and sequence comparisons. The numeri cal cutoffs are described in full on the CATH web site*. In certain dense regions of fold space (such as in the 3-layer a(3a sandwich architecture) more strict thresholds were required to subdivide topologies into smaller and more informative groupings. Only through consultation with users and a deeper understanding of fold evolution will classifications of protein structures be completely ‘error-free’.
H om ologu es, analogues and ev o lu tio n
As a result of classification efforts, two main terms are used to describe pairs of protein structures with similar folds:
• Homologous: same fold, same or similar function, common ancestry.
• Analogous: same fold, different function, ancestral origin unknown.
Since homology is defined by common ancestry it is not a difficult concept to take hold of. However ‘common ancestry’ does not necessarily infer ‘detectable sequence sim ilarity’ (see Section 1.4.1). Correctly aligned homologues can share minimal sequence identity in the range of about 0-25% known as the ‘twilight zone’ of sequence similarity (Doolittle, 1981; Sander Sz Schneider, 1991). Pairs of analogues also align with very low sequence identity, however this is obviously not sufficient evidence to rule out evolutionary relatedness. Many proteins have been evolving for a billion or more years and their sequences have diverged beyond recognition. There has also been sufficient time for the spontaneous evolution of
Chapter 1. Introduction 29
the same folds in unrelated proteins (convergent evolution). In this context, a number of commonly occurring folds (Orengo et ai, 1994) may be ‘a ttra cto rs’ in fold space. For example, the TIM barrel topology (Farber, 1993; Orengo et ai,
1994; Reardon & Farber, 1995) has an eightfold strand-helix repeat which may have simpler folding pathways and could also be the common outcome of gene duplication events.
C A T H codes and d om ain identifiers
In the CATH database, each class, architecture, topology and homologous super family can be described by numbers of the form:
C lass 3 (mixed-o/))
A rch itectu re 3.20 (mixed-o/3, barrel)
T op ology 3.20.40 (mixed-o/), barrel, TIM barrel)
H om ologou s sup erfam ily 3.20.40.10 (l,3-/?-glucanase superfamily)
Each domain in the database is identified by a six letter code of the form IxyzA l, where ‘Ixyz’ is the PDB (Bernstein et al, 1977) identifier, ‘A ’ is the chain identifier, and ‘1’ is the domain identifier. When no chain is specified in the PDB file, ‘0’ is used. A ‘0’ domain implies th a t the protein chain in question has a single domain.
1.3
M olecular interactions o f proteins
1.3.1 P rotein surfaces and binding
Chapter 1. Introduction 30
these factors, particularly in protein-protein interactions, is a m atter of contro versy (Chothia & Janin, 1975). A brief summary of the most im portant concepts is presented below.
H yd rop h ob ic in teraction s
As in the folding of protein structures, hydrophobicity is also one of the m ajor forces in ligand recognition. As the hydrophobic side-chains of a folding chain, or two interacting molecules come together there is a favourable increase in the entropy of the system as the solvent (water) molecules which were previously in an ordered shell around the exposed hydrophobic surface become disordered. There is also an energetic contribution as unfavourable apolar-polar interactions are replaced with more favourable homotypic interactions. Thus the recognition surfaces of proteins and ligands are often more hydrophobic than ‘normal’ protein surfaces (Chothia & Janin, 1975; S. Jones & Thornton, 1996).
E le c tr o sta tic in teraction s
Hydrogen bonds, salt bridges and van der Waals also provide attractive forces between molecules. Hydrogen bonds between protein and ligand can be more favourable than between protein and solvent (Fersht, 1987), and although they are not found in large numbers in all interfaces, they make an im portant contribution to the binding energy of association. Hydrogen bonds via water molecules trapped in the interface are also commonly observed. Hydrogen bonds and salt-bridges are thought to confer specificity to interactions due to their dependence on the precise location of participating atoms (Fersht, 1984; Fersht, 1987).
Chapter 1. Introduction 31
C o m p lem en tarity
Complementarity at protein-Iigand interfaces is observed in both electrostatic dis tributions (see above) and in three-dimensional shape. Shape complementarity has been quantified indirectly in terms of buried (solvent inaccessible) surface area (Chothia, 1976), gap volume (Jones & Thornton, 1995) and atomic packing den sity (Richards, 1974; Chothia & Janin, 1975; Hubbard & Argos, 1994). Recently, more direct methods have been developed (Lawrence & Colman, 1993; Norel et ai,
1994) which compare computer generated molecular surfaces. Many methods have been developed as part of docking algorithms which are described below.
1.3.2
M olecular surfaces
The surfaces of proteins are usually defined by the abstract process of rolling a hypothetical spherical water molecule over the van der Waals surface of a protein as illustrated in Figure 1.2. Three usable definitions result: accessible and contact surfaces (Lee & Richards, 1971) and the molecular surface (Connolly, 1983).
1.3.3
D ocking
The technique known as docking applies knowledge of protein surfaces and inter actions to predict the location, orientation and conformation of ligands in their physiological interactions with proteins. This is a huge field with many methods currently in development. The principles and methods of docking are reviewed in Bamborough and Cohen (1996). The extent of its success is variable and has not been rigorously tested (see Section 1.4.5, however). One m ajor problem is pre dicting the conformational changes in protein and ligand which are often observed on binding through crystallographic studies, a problem not far removed from ab
initio structure prediction described in the following section. Furthermore, the
Cliciptcr 1. I n t r o d u c t i o n 32
Probe ‘atom’
Protein atom
Accessible surface (probe centre locus)
Contact Surface Reentrant Surface
\ Molecular surface points
• Contact + Reentrant
F igure 1.2: D efin ition s o f m olecular surfaces. A d ap ted from C on n olly (1996).
1.4
Prediction of protein structure and function
1.4.1 Sequence m ethods
As the technology of DNA sequence determination continues to advance, the
amount of available protein sequence information grows rapidly. Novel sequences
result both from directed research into particular systems, and wholesale genome
and chromosome sequencing projects. In the former case, the function of novel
proteins can often be deduced from the experimental context and/or careful ap
plication of sequence database searches to identify similar sequences with known
function. Large-scale sequencing projects produce too much data for this kind of
approach. Hence automated methods are required to perform database searches
and sequence annotation in the absence of background knowledge and
conimon-sense reasoning. Accuracy and coverage are the critical performance issues for such
Chapter 1. Introduction 33
Single sequence searches
The current, widely used ‘standard sequence search techniques’ referred to in this thesis: BLAST (Altschul et ai, 1990), FASTA (Pearson, 1990) and Smith W ater man (Smith & W aterman, 1981) searches of sequence databases have very good accuracy when used with care. However, when applied to the complete proteomes
of Haemophilus influenzae, Mycoplasma genitalium and Methanococcus jannaschii
(Fleischmann et ai, 1995; Fraser et ai, 1995; Bult et ai, 1996), these methods (con fidently) find similar sequences of known function in current sequence databases for only 58%, 79% and 78% of the sequences respectively; in other words the chances of standard sequence methods failing to find a significantly similar sequence with known function for a randomly selected gene from organisms like these are around 20-40%. Does this mean th at 20-40% of proteins have hitherto unknown functions? Structural comparisons and manual sequence alignments of proteins with similar functions have identified many pairs of proteins which exhibit much lower sequence identity than the limit of around 30% above which standard sequence methods can reliably detect relationships (Levitt & Chothia, 1976; Orengo et ai, 1994). It is estim ated th at only 15% of these evolutionary relationships can be detected with the Smith W aterman single sequence method (Brenner, 1996; Hubbard, 1997). In creasing the coverage of sequence search methods to allow inferences of function from currently undetectable similarities is the goal of sequence and structure based methods (as discussed below).
M u ltip le sequence searches
un-Chapter 1. Introduction 34
known function may be used to search a sequence database (Barton, 1990; Krogh
et al., 1994, for example).
In general these methods replace the amino acid substitution m atrix used at all sequence positions with ‘position specific scoring matrices’ (PSSMs) whose scores are calculated from a single column in a multiple sequence alignment. In most cases these methods perform best where the alignment is most certain (in the more conserved regions). BLOCKS and PRINTS only scan query sequences against conserved ungapped blocks of multiple alignments. It has been shown recently th at simple embedding of consensus sequences from conserved regions of a multiple sequence alignment into a single representative sequence improves BLAST and FASTA searches, and outperforms PSSM based methods (Henikoff & Henikoff, 1997). In order to align whole sequences, gap penalties can also be calculated on a position specific basis (Gribskov et ai, 1990) and incorporated into PSSMs. Hidden Markov models (HMMs) similarly deal with position specific substitutions and gap penalties in the alignment of multiple sequences (Krogh et ai, 1994; Eddy, 1996).
Sequence co m p o sitio n approaches
Chapter 1. Introduction 35
1.4.2
Fold recognition
W ith the observation th at the number of distinct structures was not growing as fast as the PDB as a whole, it was suggested a few years ago th a t only a finite and relatively small number of fold topologies were encoded by the millions of protein sequences in nature (Chothia, 1992; Blundell & Johnson, 1993; Orengo et al, 1994). Most estimates for this limit are in the range of one thousand, and the time scale for reaching it in the order of tens of years. Bowie and coworkers realised th a t structural information could be used in an analogous way to multiple sequence information in profile methods (Bowie et ai, 1990; Bowie et al, 1991). Through the alignment and scoring of query sequences against the complete li brary of folds, the difficult problem of structure prediction by ab initio methods (discussed below) would be bypassed. W hilst the fold universe is not yet fully explored, estimates from the deposition of structures to the PDB set the probabil ity of a newly sequenced protein (with no detectable structural homologue) being similar to a known fold at 70% (Orengo et ai, 1994). Since protein structure and function, are more conserved than protein sequence, the identification of corre spondences between novel sequences and known structures would greatly assist in the characterisation of these sequences. A huge field, known as fold recognition, has developed to tackle this problem; its m ajor developments are reviewed below.
3D -1D m eth o d s
Chapter 1. Introduction 36
of alternate amino acid substitutions in the library structure. An earlier attem pt at side-chain replacement for fold recognition was too rigid to detect all but very similar proteins (Ponder & Richards, 1987).
Another algorithm developed by Bowie et al. (1990) transformed query se quences into strings of characters representing three levels of conserved hydropho bicity. Library structures were likewise converted into strings representing three levels of solvent accessibility. Dynamic programming alignments using a 3 x 3 substitution m atrix derived from database counts were able to detect remote ho mologies. More recently a large number of methods have followed this paradigm (Fischer & Eisenberg, 1996; Defay & Cohen, 1996; Hubbard & Park, 1995; Rice & Eisenberg, 1997; Rost et al., 1997); most have included secondary structure prediction information (see below) for the query sequence.
T hreading m eth od s
The 3D-1D methods described above perform respectably well despite a major lim itation in their methodology. By defining structural environments or residue classes much of the structural context information is lost, compromising the speci ficity of sequence-structure matches. In other words, one amphipathic helix or strand is much like any other, and their mis-alignment is inevitable unless more detailed structural relationships are considered. The first solution to this problem was provided by Jones et al. (1992), who applied the double dynamic programming algorithm of Taylor and Orengo (Taylor & Orengo, 1989, see the SSAP algorithm discussed above). A low level alignment is used to score the pairwise residue interactions (structural environment) at each equivalence S ij by minimising an empirical pairwise distance potential (Sippl, 1990). In other words, the low level alignment attem pts to place query residues k at positions in the library structure
I such th at the distances between j and I are similar to those observed frequently in a database for the amino acids of types i and k. As with the SSAP algorithm, the final alignment and score is traced from a high level m atrix derived from the low level alignment scores.
Chapter 1. Introduction 37
sequence in 3D as it is threaded through a library structure. The term is also frequently applied to 3D-ID methods however. Through the flexible optimisation of relative distributions of amino acids, threading methods ought to be able to detect the more distant homologues and analogues than the more rigid 3D-ID methods. In reality, however, neither m ethod has a vastly superior success rate.
The alignment problem in threading is a major one. The number of all possible sub-alignments at each equivalence is exponential with respect to sequence length (Lathrop, 1994). The double dynamic programming approach of Jones et al. (1992) does not exhaustively search this space, but is still computationally expensive. Time-consuming algorithms cannot be thoroughly tested or optimised and as a result their understanding is compromised. The ‘frozen approxim ation’ (Flockner
et ai, 1995, for example) speeds up the alignment process by testing the suitability
of pairwise distances between query residue k and library residues I rather than query residues mounted onto the structure. The residue identities of the library structure can be updated with those of the query sequence at successive iterations of the alignment (Godzik et ai, 1992). O ther algorithms to search for the best alignment have also been employed; branch-and-bound (Lathrop & Smith, 1996), Monte Carlo (Madej et ai, 1995) and exhaustive searches using heuristics (Russell
et ai, 1996).
Chapter 1. Introduction 38
1.4.3
C om parative m odelling
If, using either sequence-only or structure-based fold recognition techniques, one or more sequences of known structure are found to be related to a novel sequence under investigation it is useful to build a model of the novel protein based upon known (parent) structure(s). This is called comparative modelling. Its four main stages are: alignment of the novel sequence with the parent(s) and other homologous sequences, copying the core or framework from the parent to the model, building the non-core (loop) regions into the model, and refinement of side-chain geometry and packing. The final stages overlap with ab initio methods, described below. In general, the quality (and usefulness) of a model depends upon the relatedness of novel sequence and parent structures, and also alignment accuracy. The reader is directed to a recent review covering this large subject (Sanchez Sz Sali, 1997).
1.4.4
A b i n it i o
m eth od s
If we cannot recognise th at a sequence encodes a known fold, can we predict its novel structure from basic principles? The following sections review the more venerable field of ab initio structure prediction.
Secon d ary stru ctu ral class p red ictio n
The hierarchical route to the prediction of tertiary structure starts with the pre diction of secondary structure elements (see below) an d /o r secondary structural class. Secondary structural content can be estim ated experimentally via UV circu lar dichroism spectroscopy (Woody, 1995), or secondary structure predictions, from which the class can be derived (Rost & Sander, 1994; Eisenhaber et al., 1996b). Methods which predict secondary structural class directly are of particular interest because their output might enhance the quality of linear predictions if the infor m ation used by the two methods is independent (Rost & Sander, 1994). Nishikawa and coworkers (Nishikawa & Ooi, 1982; Nishikawa et al, 1983; Nakashima et ai,
Chapter 1. Introduction 39
been made using neural networks (Muskal & Kim, 1992; Metfessel et ai, 1993; Rost & Sander, 1994) and a myriad of variations on distance measures and multivari ate analysis (Nakashima et ai, 1986; Chou, 1989; Metfessel et ai, 1993; Klein & Delisi, 1986a; Chou & Zhang, 1995; Boberg et ai, 1995; Eisenhaber et al, 1996b).
In general the reported accuracy of these methods is around 70-80% using differently constructed datasets with varying degrees of cross-validation. Using weighted vector components or methods th a t take into account correlations be tween amino acid frequencies (component coupled) Zhang and coworkers (Zhou et ai, 1992; Chou & Zhang, 1994; Chou & Zhang, 1995) have reported accuracies at or very close to 1 0 0%, however the authors did not appreciate the memorisation effects of these param eter rich methods and neglected to perform adequate cross- validation. Eisenhaber et al (1996a; 1996b) have used a vector decomposition method to predict secondary structural content and class. Their studies showed th at dataset size, class definition and cross-validation were of critical importance to the true accuracy of class prediction from amino acid composition, which they estim ated at 60%. They showed also th a t component coupling techniques give only a small improvement in cross-validated prediction accuracy.
P r ed ic tio n o f sub-cellu lar lo ca tio n
Chapter 1. Introduction 40
of five classes: integral membrane, anchored membrane, extracellular, intracellular and nuclear. Both groups suggest physical justifications for the observed amino acid preferences. For example, the low content of hydrophobic and charged residues in extracellular proteins might allow faster transport across the endoplasmic retic ulum membrane during synthesis.
These predictions are partly academic, however, since many proteins are tagged with specific signal sequences which specify their subcellular destination, and trans membrane helices can be predicted with good accuracy (Rost et ai, 1995, and many others). Not all targeting signals have been fully characterised, however, and the membrane topology of proteins often has to be deduced from sequence analysis.
It is im portant to remember th a t large sets of structures and sequences used to test structure prediction algorithms effectively often contain proteins which fold or function in very different environments.
S econdary stru ctu re p red ictio n
The ab initio methods discussed so far have predicted only global structural fea
tures. The next level in the hierarchy of protein structure is the description of the secondary structure of each residue. Currently this can be predicted with at best about 70-75% per-residue accuracy into three states: helix, strand, coil (the Qs
measure) using multiple sequence alignments as input, making it a widely used tool in the analysis of novel sequences. Secondary structure predictions have been used in fold recognition (discussed above), in the manual assembly of tertiary struc ture models, and in folding simulations (see below). Methods to predict secondary structure have been in development for several decades and consequently the lit erature is extensive. Here, its history is briefly summarised and the most widely used methods and their lim itations are discussed.
Chapter 1. Introduction 41
1974; G am ier et ai, 1978) and complex manual analyses of patterns of physico chemical side-chain properties (Lim, 1974a; Lim, 1974b).
In intervening years, the increased availability of structural information has en abled the analysis of sequence/ structure correlations for pairs (or more) of amino acids (Gibrat et al, 1987; Rooman & Wodak, 1991; Han & Baker, 1995; Han & Baker, 1996). But their predictive power is not sufficient to make any overall improvement to the overall accuracy of secondary structure prediction. The sec ondary structure of a particular residue is partly determined by its local sequence environment, but also by its environment with respect to the rest of the folded (or folding) protein which may be separated by tens or hundreds of residues along the chain (Kabsch & Sander, 1984).
Although Robson and colleagues (Gam ier et ai, 1978) recognised th a t aligned protein sequences would provide valuable evolutionary information relevant to sec ondary structure prediction using their algorithm (known as the GOR method), it was not until relatively recently th a t the databases contained enough related sequences to put this into practice (Zvelebil et ai, 1987). The improvement was a significant one, many workers have repeated the experiment with a variety of algo rithm s, and in each case a 5-10% increase in was reported. Multiple sequence alignments provide information about core secondary structures through the con servation of amino acids and the location of insertions and deletions. Sequence conservation also highlights patterns of key physico-chemical features which relate to secondary structures, reducing the noise problem in single sequences.