INFANTILE AMAUROTIC FAMILY IDIOCY
Occurrence, Gene-ticConsiderationsand Pathophysiology
in the Non-JewishInfant
Stanley M. Aronson, M.D., Marius P. Valsamis, M.D., and Bruno W. Volk, M.D.
Isaac Albert Research Institute of the Jewish Chronic Disease Hospital, Brooklyn, and the Departments of Pathology, State University of New York, College of Medicine, Brooklyn, and the
Albert Einstein College of Medicine, Bronx, New York
T HE ETHNIC spectrum of infantile amau rotic family idiocy has been progres sively broadened as further cases have be come available for study. The existence of the disease in European and American chil dren of non-Jewish background has been repeatedly recorded;126 cases have also been described in children of Asiatic and Levantine origin.27-33 The initially con sidered doctrine that the disorder is con fined exclusively to infants of Jewish herit age is therefore no longer tenable.
The present report adds brief data on six
additional non-Jewish Caucasian infants
with infantile amaurotic family idiocy, as well as more extended observations regard ing the clinical, biochemical and pathologic findings in two Negro infants with this dis order. Limited clinical information regard ing a third Negro infant with this disease is also included. These latter cases constitute the first known description of the disease in the Negro race. Information concerning 79 cases of infantile amaurotic family idiocy in the non-Jewish infant, derived from pre viously published reports, has been com bined with the nine presently reported cases. The amalgamated data are contrasted with the findings derived from 148 per sonally collated records of Jewish infants in whom the disease was verified, to deter mine whether ethnic factors appreciably in fluence the patterns of genetic transmissi bility, the degree of abnormalities in bio logic fluids, the clinical onset and progres sion, and the underlying pathologic changes inherent in the disease.
CASE REPORTS Case 29*
This Negro male infant was born spon
taneously at full-term, with no complications
of pregnancy or labor recorded. The parents
had one previous child, a healthy male. Birth weight of the present infant was 3.43 kg; head circumference, 38.1 cm. Some transient cyano sis was noted during the first postpartum day, but this difficulty subsided shortly thereafter. The infant was discharged from the hospital on the seventh day. The parents could not recall any notable abnormalities during the first 5 months of life. At approximately the age of 5 months, however, the mother described a mild
apathy in her infant son, together with a
greater tendency to sleep.
The child was admitted to Kings County Hospital at the age of 7 months because of bronchitis. Physical examination disclosed a poorly developed and retarded infant with stertorous respirations. The child could not sit up, and did not follow light or recognize objects. Bilateral inguinal hernias were noted. The head circumference was increased (47.8 cm) and the anterior fontanelle measured 8.9 cm. A bilateral convergent strabismus and
lateral nystagmus were apparent. Pupils re
acted well to light. No funduscopy was per formed. A palatal deformity and an unusually enlarged uvula were evident. The child was discharged at the age of 8 months.
The infant was readmitted at the age of 14 months because of fever, anorexia and further retardation of development. He was described as lying quietly in bed, exhibiting only a feeble cry. -The tendon reflexes were severely
* Case numbers referred to in the present paper
represent the series number of such cases in a reg istry of central nervous system lipidoses.
Supported in part by a grant from the National Tay-Sachs Association.
ADDRESS:(B.W.V.) Isaac Albert Research Institute, 86 East 49th Street, Brooklyn 3, New York.
diminished or abolished and a bilateral, tran
sient ankle clonus was present. The head was enlarged to 49.5 cm and the chest measured 42 cm in circumference. A lumbar puncture was performed and showed no abnormalities of the cerebrospinal fluid. A month after ad
mission, at the age of 15 months, the infant died.
No history of consanguinity could be estab lished in extensive conversation with the par ents. Both mother and father were Negro and both were born in small, geographically sep arated communities of South Carolina. There was no suggestion of any parallel illness in more remote relatives, nor was there evident participation of any ethnic groups other than Negro in the family lineages.
NECROPSY FINDINGs: Pertinent findings were confined to the nervous system.
Gross Examination. The brain weighed
1,060 gm. The cerebral hemispheres were
normal in contour and were symmetric. The dorsal gyri, bilaterally, were unduly prominent
and the intervening sulci widened. The
leptomeninges were unremarkable except for
congestion of the cerebral veins. The vessels at the base of the brain were of normal dis tribution and caliber. The brain stem and cerebellum were of slightly diminished vol ume. Dissection of the supratentorial tissues disclosed a mild dilatation of the lateral ven tricles. The cortical mantle was grossly unre markable. The bilateral cerebral white mat ter showed an apparent loss of myelin, this change most marked in the parietal and occipi tal lobes. Dissection of the brain stem and cerebellum showed no gross abnormalities be yond the mild atrophy previously noted.
Microscopic Examination. Histologic sec tions derived from all levels of the neuraxis showed a ubiquitous neuronal involvement. In the cerebral cortex there was a perceptible reduction in the number of demonstrable neurons and a severe degree of enlargement of those remaining. The typically involved cell was ballooned and the cytoplasm filled with a granular, poorly staining substance which was faintly sudanophilic and stained positively with the periodic acid-Schiff reaction. The gangli onic nuclei were deviated eccentrically and the Nissi substance was greatly diminished or absent. Large numbers of activated microglial cells, filled with sudanophilic material, were noted throughout the cortex, often congregat
ing about small blood vessels. Myelin stains of
the subjacent white matter showed a moderate loss of myelin and appropriate stains showed a comparable loss of axons. A diffuse astrocyto
sis was apparent in the cerebral white matter,
somewhat more perceptible in the frontal lobes. Sections of the basal ganglia showed comparable, but less marked, changes within the contained neurons. A microgliosis was
again evident.
Sections at various levels of the midbrain,
pons, medulla and spinal cord also demon strated the diffuse neuronal lipoid accumula tion. A quantitative distinction, however,
could be demonstrated, i.e., the tegmental
neurons showed the greatest degree of in volvement, while the inferior olivary nuclei demonstrated the least degree of cytoplasmic storage. Sections of the cerebellum showed only a mild degree of Purkinje cell involve
ment and an atrophy of the internal granular
layer cells. Ganglion cells of the peripheral autonomic nuclei also showed ballooning de generation.
Case 157
A full-term Negro female infant was born after an uneventful pregnancy and labor. Birth
weight was 3.2 kg. During the first months of
life, the infant developed in an apparently
normal fashion accompanied by normal weight
gain. No feeding problems were encountered. At the age of 6 months, the infant was able
to sit unsupported and was capable of grasping
and picking up objects. At the age of 8 months, the parents noted lassitude and failing vision in their infant. Because of the latter disturb ance, she was brought to another hospital.
Physical examination at the age of 9 months
disclosed bilateral retinal cherry-red spots. Within the succeeding few months, the child
displayed periodic convulsive movements as
well as a severe hyperacusis. Progressive pare
sis also developed during this period. At the
age of 23 months, following multiple hospital
izations, the child succumbed.
infant was born in North Carolina and the pa
ternal grandfather in South Carolina. There
was no information to indicate any consan guinity. The patient's mother was horn in New
York State and her father and mother were
born in North Carolina and New York City, respectively. No evidence of consanguinity on the maternal side and no neurologic disorders in any of the maternal relatives were elicited. The patient represented the fourth preg
nanc@ of this marriage. The first child, a full
term male weighing 3.6 kg at birth, was con sidered normal until the age of approximately 18 months. At this time the parents were con cerned because the child showed no tendency to walk and was unable to stand without sup port. Coincidentally, the child showed an un usual sensitivity to loud noises and had diffi culty seeing. Because of the associated, progressive paresis and failure to eat properly,
the child was admitted to another institution
where, at the age of 33@\ears, he died. A
necropsy was performed at another hospital and the histologic findings were consistent with a diagnosis of infantile amaurotic family idiocy. The second and third pregnancies
ended prematurely in miscarriages.
NECROPSY FINDINGS: Gross Examination.
The brain weighed 980 gm. The cerebral hemispheres were symmetric and of normal
external contour. The individual gyri ap
peared to be somewhat more voluminous than
normal. No externally demonstrable lesions could he seen and the leptomeninges were grossly unremarkable. In contrast to the
slightly increased volume of the supratentorial
tissues, the cerebellum was distinctly atrophic and a disparity between supratentorial and infratentorial tissues was striking (Fig. 1). Consecutive cross sections through the supra
tentorial tissues disclosed a moderate degree
of demyelination deep within the white matter without any coexistent breakdown in tissue. The line of demarcation between cortical gray
matter and underlying white matter was ill defined. The ventricular cavities were mildly
compressed. No focal lesions were macro
scopically demonstrable. Dissection of the
brain stem and cerebellum showed no abnor
malities except for a distinct and diffuse atrophy
of the cerebellar cortices. The individual folia
were diffusely shrunken and the interfoliar spaces unusually widened. The density of the tissues comprising the cerebellar hemispheres was distinctly increased.
Microscopic Examination. Representative
blocks of tissue, derived from all levels of the neuraxis, were taken for histologic assessment
and stained by a wide range of histologic pro
cedures. Large numbers of sections derived
Fic. 1. Lateral view of brain (Case 157). Note the relative atrophy of in
232
‘¿5 @- - - •¿@@@
- _‘@ @5 S_. .@
-. - •¿5@- —¿
•¿ 5@__#
- @_-•_-._---
@--.
@ -.-@-:.@“.
- @.‘:‘ - - - —¿
-•¿ -•• ‘¿
@ ;
--‘¿I,
0@
a -‘
a
@‘¿
s@, @
FIG. 2. Microscopic view of tegmental neurons of medulla (Case 157) show
mg marked cytoplasmic ballooning, reduction of chromatin content and mild reactive gliosis. (Hematoxylin and eosin, x 150.)
inferior olivary nucleus of the medulla. These latter cells showed only a mild degree of neuro cellular distension.
Preparations of cerebellar cortex showed a moderately severe diminution in the number of cells within the internal granular layer but flO reactive astrocytosis. The Purkinje cells were minimally' decreased in number and showed only a mild degree of cell ballooning. Stains for neurofibrils demonstrated a mild re
duction in these intracellular structures as
well as an atrophy' of the dendritic projections.
No axonal ballooning was demonstrable. With
in the cerebehlar white matter, there was noted a diffuse decrease in mvelin content, quantita tively' more severe than that displayed in the cerebral white matter. The deep paraventricu lar nuclei of the cerebellum showed minimal dystrophic changes quantitatively comparable to those noted in the inferior olivary nuclei. Rarely, collections of activated microglia filled with sudanophihic material were seen in the surrounding interstices.
BIOCHEMICAL OBSERVATIONS (CASE 157)
Sustained and significantly increased ac tivity of glutamic-oxalacetic transaminase and lactic dehydrogenase have been de
scribed in the serum and cerebrospinal fluid of infants with infantile amaurotic family
from the cerebral cortex showed a uniformly severe ganglion cell enlargement (Fig. 2). The nuclei were invariably displaced to the pe riphery of the cell and the Nissi substance was diminished or absent. The increased vol ume of c@-toplasm was cofllpOsed of a mildly sudanophilic, slightly granular substance which showed minimal staining with hema
toxvhin-eosin. While all cortical neurons were
demonstrably involved in this process of in tracellular storage, the number of such cells
showing neurocytolysis was minimal. Only a
slight reactive microgliosis and no appreciable astrocytosis was noted in the cortex.
Sections taken from underlying white mat ter, however, showed a diffuse pallor with
myehin stains, a mild loss of axons, but no significant ghiogenous response. Sections from
basal ganglia showed similar but quantita tively' subdued changes within the ganglionic c@'toplasm. The cell population in the deep
gray matter was noticeably decreased. The
ependymal lining cells showed no abnormali ties.
Case
157Other Cases*Normal RangeSerumGlutamic-oxalacetictransaminase139*597@—
40Lactic
dehydrogenase1,4161,384@OO—68OAldolase10.710.85—
10Cerebrospinal
FluidGlutamic-oxalacetictransaminase66384—
14Lactic
dehydrogenase9110@5—
40Aldolase1.50-1
Glutamw Oxalacetic
Transaminase
(%)*Lactic
Dehydrogenase
(%)*Cerebral
gray matter5158Cerebral
white matter667@Brain
stem4764Cerebellum6780
tissue were assayed for activity of glutamic
oxalacetic transaminase and lactic acid de hydrogenase. A striking decrease in tissue enzyme activity was evident in the cerebral
cortical gray matter and to a slightly lesser
degree, the cerebral white matter. In con trast, there was only a moderate decrease of lactic acid dehydrogenase activity in the pontine and cerebellar tissues (Table II). In addition to these biochemical deter minations, an additional segment of pontine tegmentum was used for determination of tissue neuraminic acid.36 An approximately threefold increase of neuraminic acid was evident in the tissues of Case 157 as con trasted to control tissues obtained from in dividuals dying of nonneurologic disease.
Case 30
A white, male infant was born after an un eventful pregnancy. Both parents were non Jewish, and could recall no ancestors of Jew
ish faith. The father's family migrated from
France and the mother's grandparents from Sweden and France. No information indicat ing consanguinity was obtained. The first noted abnormality in the infant was a severe
reaction to loud noises, beginning at the age of 6 months. In the succeeding 6 months, the
hyperacusis became more pronounced and was accompanied by progressive apathy, pa ralysis and failing vision. Examination of the
fundi disclosed bilateral cherry-red spots. The
child died at the age of 25 months. Necropsy
was not performed.
TABLE II
LEVELS OF ENZYME ACTIVITY IN BRAIN
TISSUE FROM CASE 157
TABLE I
BIocIIEMmc@u@ I)ATA IN CASE 157
* Data determined from 19 patients with confirmed
infantile amaurotic family idiocy. Only values derived
from patients @0to Q4 months ofage are included. ** All values expressed in units/ml of serum or
cerebrospinal fluid.
idiocy.@@'@@The increment in enzyme ac tivity in these biologic fluids is most striking in the initial months of the disorder. Only after the second or third year do the values for activity of these enzymes approach the normal range. Aldolase activity in cerebro spinal fluid pursues an approximately paral lel course. Activity of aldolase in serum, on the other hand, tends to be elevated coin cident with the emerging atrophy of skele tal muscle characteristic of the disease. Table I lists the findings of the three en
zyme procedures employed in Case 157.
These abnormal values are contrasted with comparable determinations in 19 Jewish in fants with infantile amaurotic family idiocy. Since the extent of enzymatic variance dif fers with the stage of the disease, only values obtained between the ages of 20 to
24 months are included in the second
group. It is seen that the abnormal values obtained in Case 157 are in accordance with the alterations previously noted in this dis order.
Necropsy of Case 157 was performed
within 5 hours after death. In addition to tissues employed for histologic study, rep resentative blocks of fresh nervous system
* Data expressed as percentage of normal (the latter
values obtained from brain tissues of infants dying of
234
birth, was not a prominent clinical feature until the child was 8 months old. At the age
of 1 year, blindness and convulsions super vened, and cherry-red spots were observed in
both optic fundi. The child regressed further
to a vegetative state by 18 months of age and
died at 46 months. No necropsy was per formed. A younger sister developed normally
and is alive and healthy.
Case 147
This white, male infant was born after a
full-term pregnancy. The ancestors of both parents were born in Germany and were non
Jewish. No information to indicate consanguin ity was obtained. The infant's development
proceeded normally, but at 8 months of age
some weakness and paucity of spontaneous
activity were noted. Hyperacusis became
prominent at 11 months, and convulsive epi
sodes commenced at the age of 14 months.
The child's vision at this time was difficult to
evaluate, but some sight was preserved.
Ophthalmoscopic examination showed the
existence of bilateral, retinal cherry-red spots and diffuse macular pallor. The child was alive at age 16 months. A younger male sibling is healthy and has developed normally.
DISCUSSION
The initially reported descriptions of in fantile amaurotic family idiocy were con fined exclusively to children of Jewish ex traction, generally of Polish-Russian origin. In 1901, 14 years after the inaugural de scription of the disease by Sachs, Falken heim1 gathered 36 cases of the disorder, noting that 4 of these children were of non Jewish origin. His review, however, made no further reference to these latter cases nor gave the criteria for inclusion as sub stantiated instances of the disease. Subse quent surveys by other authors also made vague referral to cases of non-Jewish an cestry, but such reports were equally in complete. The overwhelming preponder ance of the disease in Jewish infants and the lack of authentic verification in those rare reports of the occurrence of this mal ady in children of other ethnic backgrounds has prompted Sachs to state in 1910 that, in Case 31
A white,femaleinfant,the youngersister of Case 30, was spontaneouslydelivered
after a 37-week pregnancy. Development was seemingly normal until the age of 6 months, when abnormal eye movements and hyperacu sis ensued. Increasing irritability, weakness and blindness began shortly thereafter. Ret
inal examination showed the presence of bi
lateral cherry-red spots. The infant was alive at the age of 13 months.
Case 117
The patient was a white, female infant, the first-born child of a father with Norwegian,
non-Jewish ancestry. The mother's grand parents were born in Germany. The child's maternal greatgrandmother was Jewish, but all other members of the family tree were of non-Jewish extraction. No evidence of inter marriage was recorded. The pregnancy and
delivery of this child were normal. At the age
of 4 months she could not hold her head up.
Shortly thereafter she showed weakness, irrita
bility and blindness. Examination of the optic fundi at the age of 5 months disclosed cherry red spots in both eyes. Despite severe paraly
sis and inanition, the child was still alive at
age 25 months.
Case 118
This male child was the younger brother of
Case 117. He was born after a gestation of 9
months, and delivery was uncomplicated. At
2 months of age the child displayed weakness,
convulsions and arrested development. Blind ness became apparent a few months later, and the child died at the age of 6 months. Bilateral cherry-red spots in the retina were recorded shortly before death. No necropsy was performed.
Case 140
The patient was a white, female infant. The father's parents derived from Guatemala and Italy. The mother's parents were born in mid western United States. None of the infant's forebears were Jewish. There was no history
of consanguinity. The child developed nor
No. Cases Reference No. his opinion, “¿theinfantile form invariably
occurs among Hebrews and among them
only.―37
The first adequately described report of infantile amaurotic family idiocy in a non Jewish infant published in 1914,2 was a male infant of British antecedence, healthy until 5 months of age and subsequently showing progressive limb weakness, dis interest in his surroundings and, upon oph thalmologic examination, exhibiting bi lateral cherry-red spots. In 1916, Tarr27 de scribed a female child of non-Jewish Syrian precursors, who showed the classic symp toms of this disorder and bilateral cherry red spots. The first report of necropsy con firmation, published in 1930,12 was that of a white male infant, of Irish-German de
scent, who died at the age of about 16
months. The necropsy findings were clas sically those of infantile amaurotic fam
ily idiocy. Subsequently, necropsy findings
of 14 additional cases have been pub
lished@*_1Y,bO,20,23-26,31 In one such report,
neuraminic acid levels were shown to be significantly elevated in cerebral and cere bellar tissues.19
In 1923, Sachs6 acknowledged the exist ence of the disease in infants of non-Jewish extraction and described two children of separate sibships with this disorder who had come under his purview. In the suc ceeding decades the disease was recorded in non-Jewish children originating from all continents. Over 100 separate instances of the disease with such antecedence have been described in the medical literature. The geographic sites of origin of the ma jority of these reports are summarized in Table III. A significant number of the non Jewish examples of the disorder appeared to originate in the British Isles and in Swit zerland. This seeming geographic concen tration of non-Jewish cases probably re flected a greater measure of medical obser vation and has not been construed as having the same genetic significance as the pre ponderance of Jewish cases originating from the northeast territories of Europe.38'39
TABLE III
SITES OF BIRTH OF 88 PATIENTS WITH INFANTILE
AMAUROTIC FAMILY IDIOCY
10 2,14,16,20
1 22
23 23
4 1,3,19
4 21
2 13
3 32
1 30
3 33
2 31
6,7,9,15 5,26,S@5
17,S
18 S S
28 27
12,24 4,11.25,S
Europe
England
France
Switzerland Germany
Norway Spain
A@,ia
Japan
China India
Ceylon
Africa
Egypt 5 29
North America, Australia*
BritishIsles 4
Italy 5
Norway 3
Finland 1
France 2
Germany
Japan 3
Syria
West Europe, mixed 3
Unspecified 7
88
Total
* In instances where the infant was born in North
America or Australia, site of birth of parents is listed.
@ Presentseries.
Genetic Observations
Most observers have recognized the diffi culties involved in proposing comparative incidence rates of infantile amaurotic family
idiocy in different ethnic divisions. The de
gree of ascertainment differs appreciably in various geographic locations. Reliance upon published reports, on the other hand, is equally hazardous since the more unusual case (e.g., of non-Jewish background) is the more likely to emerge in the medical
literature, thus introducing a bias into the
cumulative statistics.
of this disease in large urban populations. An analysis of 36,000 consecutive pediatric
admissions to a large Chicago hospital re vealed 12 cases of infantile amaurotic fam ily idiocy in the 17-year period covered by
this survey.40 One of the 12 cases was an infant of non-Jewish background. However, that study does not indicate the religious substrate of these 36,000 admissions so that no comparative rate can be constructed.
A more comprehensive survey covering a
period of 10 years and based upon exami nation of death certificates in New York City, was carried out by Kozinn et al.@' The derived data, projected against a background of known numbers of births of the various religious backgrounds, permitted these workers to propose a carrier (heterozygos ity) rate for this urban area. On the basis of their findings these authors estimated the rate amongst Jews to be approximately 1:45. A rate of about 1:333 has been sug gested for non-Jews in this same region. Parenthetically, these authors have postu lated that the total number of cases of the disorder throughout the world, occurring in non-Jewish children, is theoretically in ex cess of those in Jewish infants. Hanhart's meticulous study23 of the disease in Switzer land, in actuality, has uncovered 33 clini cally verified instances of the disorder, and a majority of these were reputedly of non Jewish origin.
The scope of description in previous re ports of infantile amaurotic family idiocy in non-Jewish children is variable and only a small fraction of the cases thus recorded were ultimately verified by histologic ex amination of the nervous system tissues. In the absence of such morphologic confirma tion the following criteria were used by the
presentauthorspriorto acceptanceof the
case as an instance of the disease: 1) Onset
of symptoms prior to the age of 18 months of life; 2) clinical symptoms, including pro gressive apathy, paresis and amaurosis; 3) bilateral cherry-red spots. In some instances, an infant fulfilling the above qualifications was described by an observer and in further evaluation of the family, previous siblings
with a comparable disease pattern were belatedly described by the parents. How ever, in such instances examination of the fundi was obviously not performed. Where one sibling was known to have had the dis order, the tentative assumption was made that such siblings were probably afflicted with the same disease. On this basis 88 cases of the disease in non-Jewish children (including the nine presently reported in stances of the disease) were analyzed.
While the existence of infantile amau rotic family idiocy in siblings born prior to the propositus case is presently accepted
where proposed by previously authors,
some reservations must be entertained. The reports of Hanhart23 and Mathur and Sri vastava33 each describe the occurrence of the disease in three out of three siblings. Nordlow published data2' concerning a non Jewish Scandinavian sibship composed of four children, all of whom were afflicted. Some of the other previously published re ports also indicate an unusually high intra
familial incidence.23'29 Review of the com
bined data derived from 124 Jewish pedi grees with proven infantile amaurotic fam ily idiocy discloses 33 three-child families,
none of which contain more than two
instances of the disorder. The statistical chances of three-out-of-three or four-out-of four in a disease inherited as a Mendelian recessive are of such a low order as to suggest that one or more of the previous siblings in such families, labelled as having infantile amaurotic family idiocy, may have succumbed from a different disorder.
A factorial assessment of the non-Jewish sibships (Table IV) shows that the heredi tary transmission of the disease in the non Jewish families is nevertheless compatible with a hypothesis of a simple autosomal
recessive disturbance and, therefore, does
not differ from the patterns of genetic trans missibility exhibited by the carrier fam. ilies of Jewish extraction. The information -derived from 148 verified examples of the
disorder in children with Jewish back
A No. Children . in FamilyB No. •¿â€¢¿ FamthesC Total No. S Children (AXB)I) No. Children •¿ with IAFIF , @ I Observed °J,'* IAFI Cojculated °C** ‘¿ JAFI 124 Jewish . Szbships (%)1 2 8 4 .5 6 7 810 10 12 7 4 3 2 310 20 36 28 20 18 14 2410 13 20 14 10 7 5 9100 65.0 55.6 50() 50.0 38.9 35.7 37.5100 57.1 43.2 36.5 32.8 30.4 28.8 27.7100 61.3 42.4 31,3 30.0 —¿ —¿ 37.55117088X@=6.08
p= •¿98@ItX52=0.77 p= .54@
No. Sib lingsNo. with IAFIPer CenI Siblings with IAFIJewish (124 sibships*)Male1508959.3Female1226250.827215155.5Non-Jewish (51 sibships**)Male684363.2Female794050.6Sex unstated23521.7Total1708851.8 TABLE IV
OBSERVED AND THEORETICALLY CALCULATED NUMBER OF AFFECTED CHILDREN IN 51 NoN ,JEWIsH SIBSHIPS WITH INFANTILE AMAUROTIC FAMILY IDIOCY (IAFI)
* For method of calculation, see reference 39. @ Personal series.
t Total theoretically calculated number of patients with the disorder in total sibships of 170is 69.9 (FXC/100) compared with observed number of 88 (Column D).
¶Differencesbetween observed and expected are not significant.
The sex incidence of the disorder was originally thought to be preponderantly
female.6,28 As more cases became available for statistical inquiry, this initial impression was vitiated. The comparative rate of the disorder relative to sex in the accumulated sibships, regardless of ethnic background, shows no apparent sex linkage (Table V). While more males were afflicted with the disorder than females, the total number of males in the combined sibships exceeds the total number of females and the compara tive incidence rate in each of the sexes is approximately equal.
Examination of the family trees under consideration demonstrates one appreciable distinction relative to religious origin. Amongst the 124 pedigrees of Jewish fam ilies with infantile amaurotic family idiocy 12 (10.3%) produced information indicating the existence of this disorder in near rela tives (exclusive of siblings). No cases were described in the near relations of the series composed of 51 non-Jewish families.
Medical records concerning 751 first cousins of the 148 Jewish infants with in
fantile amaurotic family idiocy were avail able. Of these, only two were afflicted with
TABLE V
SEX INCIDENCE OF CHILDREN IN FAMILIES WITH INFANTILE AMAUROTIC FAMILY
IDIOCY (IAFI)
* Cases personally recorded by authors.
238
the same disorder (an incidence of 0.0026). A theoretic rate of such an occurrence can be ascertained by compound probability calculations. The probability of heterozy gosity for the abnormal gene among the siblings of the proband parents is 1:2 (or 0.5). The choice of marriage partner of such siblings is apparently not based on random mating, but rather confined to co religionists. Kozinn and associates4' have calculated the incidence of individuals carrying the abnormal gene in the Jewish population at 1:45 (or 0.022). The chance, therefore, of a parental sibling and his or her mate both being heterozygous for this disease will therefore be about 1:90 (or 0.011). Since the disease is a recessive dis
orderwith completepenetrance,and one of
four (or 0.25) children theoretically homo zygous for the abnormal gene, the antici pated probability of a first cousin of a Jew ish infant with infantile amaurotic family idiocy being afflicted with the same disease
becomes 1:360 (0.5 X 0.022 X 0.25 =
0.0028), which is remarkably close to the observed rate of 0.0026.
Applying the same calculation to the non-Jewish group and assuming that the carrier frequency for the disease amongst non-Jews is 1:333 (or 0.003), the calculated probability of a first cousin of an affected individual having the disease is 1:2,664 (or 0.00038). The average number of first cous ins per propositus in the well documented Jewish group with infantile amaurotic family idiocy is 6.4. If this figure is tenta tively projected to the non-Jewish family constellations, one may expect one first cousin to have the disorder in 417 non Jewish families so affected. The total num ber of non-Jewish families with the disorder summarized in the medical literature is but a fraction of this. The probability, therefore, of such an occurrence among the non-Jewish families is so remote that the non-Jewish cases appear to stand out like isolated instances in a family arborization otherwise untouched by the disease.
In this regard, however, it is interesting to note Hanhart's extensive geneologic in
vestigation of the affected pedigrees in Switzerland.23 While there was no expres sion of the disorder in immediate relatives, he was, nevertheless, able to trace a large number of his cases back through three cen tunes to one original marriage, with numer ous subsequent instances of interlocking consanguinity.
ROLE OF CoNsANcuINrrY: The factor of
intermarriage has been repeatedly empha
sized in diseases characterized by genetic passage, since inbreeding enhances the possibility of homozygosity in the prog eny.42 In previous summaries of infantile amaurotic family idiocy, the rate of con sanguinity has been estimated as high as 5Ø%@4344 An inverse relationship between gene frequency and incidence of consan guinity has been postulated. Thus, for ex ample, the more rare the carrier (heterozy gous) state, the more common will be inter marriage in the family configurations con taining the clinically apparent disorder. Cousin marriages were actually observed in 2 of 124 Jewish families studied (an inci dence of about 1.7%), while at least 30% of the reported non-Jewish pedigrees with in fantile amaurotic family idiocy involved consanguineous marriages. The expected frequency of consanguinity can be roughly calculated with Lenz' formula,45 and this proves to be 1 to 3.5% for affected Jewish families. On the other hand, in non-Jewish affected families (worldwide), an expected rate of 4-16% is calculated which is signifi cantly less than the observed frequency. This discrepancy may perhaps be due to an erroneous assumption that the heterozy gosity rate of 1:333 for this disorder (a figure derived from statistical studies per formed on a non-Jewish American popula tion) is applicable to all non-Jewish ethnic groups. If non-Jewish families reported only
from the United Kingdom and United
Onset Duration Death@on@hs
No. No. Months No.
Cases Cases Cases
—¿-1-011 1 4 2 6
1 5 1 9
12
3 7 2 10
13
2 8 3 12
24
8 9 7 13
35
9 10 3 14
36
20 11 2 15
87
3 12 3 16
28
2 13—16 6 17—20
139
3 17—20 6 21—24
1110
5 21—24 4 25—28
111
1 25—32 6 29—32
612
3 33—40 2 33—40
713
1 41—48
218
2Total
64 47*
61**Av@r
age 6.6 15.6 21.5
239
than any intrinsic factor modifying the basic velocity of the disease.
Some of the children in the non-Jewish
category failed to show macular degenera
tion despite repeated funduscopy. Some form of optic atrophy, however, was gen erally recorded. All of the Jewish children showed typical cherry-red spots. Since the lack of classic macular degeneration has been noted by others in children of all ethnic groups, in whom the diagnosis was substantiated by subsequent necropsy ex amination, it is possible that these aberrant cases are still examples of the disorder.@ In one of the accepted cases'6 alluded to above, primary optic atrophy was demon strable; this child of non-Jewish back ground eventually came to necropsy and the classic histologic changes of the dis ease were found. While macular degenera lion is at times inapparent in otherwise typical cases of this disorder, there are no substantiated reports of the presence of cherry-red spots in any disease other than infantile amaurotic family idiocy or Nie mann-Pick disease with central nervous system complication. The rare reports of comparable retinal findings in juvenile am
aurotic idiocy are believed by Wybum
Mason to be vague: “¿thedescription in most of these does not carry conviction, the
appearances were not typical and were
merely slight variations of the pigmenta tion usually occurring in the juvenile type.―2°
Pathologic Observations
The histologic studies performed on the two Negro infants with infantile amaurotic family idiocy disclosed no conclusive dis tinctions from the typical cases to justify any separation. The neurons of the nervous system including those of the autonomic ganglia showed a plenary involvement. Typically, the ganglion cells were increased in volume and showed a ballooned outline. The Nissi substance in the cytoplasm was markedly reduced in amount. It has been shown previously that a hierarchy of gan glionic implication exists which is seem TABLE VI
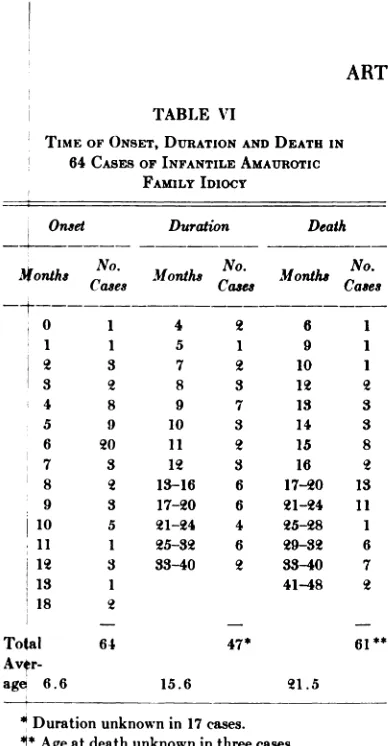
TIME OF ONSET, DURATION AND DEATH IN
64 CASES OF INFANTILE AMAUROTIC
FAMiLY IDIOCY
Durationunknown in 17 cases.
@ Age at death unknown in three cases.
CI@nicaIObservations
The age when pertinent symptoms com
m@nced was judged to be 5.9 ±0.3 months in the Jewish children afflicted with this di@ease. Amongst the non-Jewish children with infantile amaurotic family idiocy the on@et was determined at 6.6 ±0.4 months wi@h a variation ranging from birth to 18 m4nths (Table VI). The symptoms initiating pakental concern did not vary appreciably
be@tween the two groups, although the find
ingly related to the phylogenetic status of the various nerve cell groups.47 Nuclei of more recent development (e.g., neopallium) are the most severely involved while those of more archaic precedence tend to be less involved (e.g., olfactory bulbs, paleostri atum and cerebellum). As survival is pro longed, the gradation of involvement be
comes less pronounced. The two Negro
infants succumbed at ages 15 and 23
months, respectively. The degree of abnor mal lipoid deposition in the cerebellar neurons was strikingly minimal, but in
keeping with the abbreviated clinical
course. The extent of reactive gliosis, particularly in Case 157, was far less than that seen in the usual case of infantile amaurotic family idiocy, but this, too, is probably related to the abortive course. In reviewing the 12 separate publications containing pathologic data of non-Jewish cases12' 14-17,19,20,23@2631 there does not ap pear to be any evidence to indicate any singular structural features when contrasted to the classic morphologic findings in the Jewish infant with this disorder. In Case 157, in addition to the histologic techniques employed, biochemical determinations of
—¿i+90
@ +80
0
@ +70
o +60
LL. +50
2 +40
+30
@ +20
enzyme activity and neuraminic acid con
tent were also carried out. The quantita tive alterations were in complete conformity with the changes characteristic of the dis ease in the Jewish child.
A progressive enlargement of the head, generally evident beyond the age of 24 months, has been consistently observed in children of Jewish extraction with this dis order.@8 Sporadic observation of this phe nomenon has also been noted in previously reported instances of the disease in non Jewish children.30 This head enlargement has been shown to be a reflection of a pro gressive megalencephaly, rather than any obstructive hydrocephalus. While no in crease in brain weight was evident in the two Negro children described here, it can be seen in Figure 3 that their time of death preceded the usual age of development of secondary gliosis of the cerebral hemi spheres, the underlying lesion responsible for the reactive megalencephaly.
SUMMARY
The clinical, biochemical and pathologic findings regarding two Negro infants with infantile amaurotic family idiocy have been
.
.
. S
.
•¿ •¿ •¿S.S
.
w
(@+I0 S
@ 0 1 I I'°I I T I I I I I I I I I
C.) A 4 8 12 1620.02832 3640444852 56 6OAGE,
lv MONTHS
O@@2O
Fic. 3. Graph showing weights of brains of 26 infants dying with infantile amaurotic family idiocy (expressed as percentage deviation from normal) plotted against age in months. Megalencephaly emerges approximately after 2 years of life. The two Negro infants with the disease (open circles) do not show any relative increase in brain weight; however, they did not sur
1960;26;229
Pediatrics
Stanley M. Aronson, Marius P. Valsamis and Bruno W. Volk
and Pathophysiology in the Non-Jewish Infant
INFANTILE AMAUROTIC FAMILY IDIOCY: Occurrence, Genetic Considerations
Services
Updated Information &
http://pediatrics.aappublications.org/content/26/2/229
including high resolution figures, can be found at:
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtml
entirety can be found online at:
Information about reproducing this article in parts (figures, tables) or in its
Reprints
http://www.aappublications.org/site/misc/reprints.xhtml
1960;26;229
Pediatrics
Stanley M. Aronson, Marius P. Valsamis and Bruno W. Volk
and Pathophysiology in the Non-Jewish Infant
INFANTILE AMAUROTIC FAMILY IDIOCY: Occurrence, Genetic Considerations
http://pediatrics.aappublications.org/content/26/2/229
the World Wide Web at:
The online version of this article, along with updated information and services, is located on
American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.