Genome of Russian wheat aphid an economically important cereal aphid
12
0
0
Full text
Figure
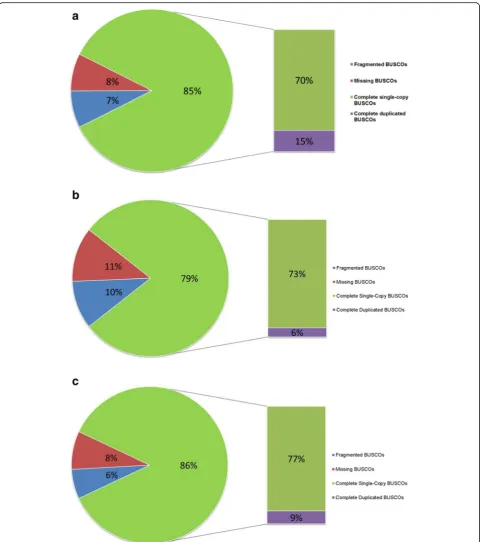
![Table 1 Classification and general features of Diuraphis noxiabiotype SAM [22]](https://thumb-us.123doks.com/thumbv2/123dok_us/647629.2064552/2.595.304.538.106.507/table-classification-general-features-diuraphis-noxiabiotype-sam.webp)

![Fig. 2 PAUP generated phylogenetic tree based on whole mitochondrial genomes. A maximum parsimony tree generated through PAUP [56]utilizing whole mitochondrial genomes, that was aligned with MAFFT [57], illustrating Diuraphis noxia’s close association with Acyrthosiphon pisum](https://thumb-us.123doks.com/thumbv2/123dok_us/647629.2064552/4.595.56.290.526.731/phylogenetic-mitochondrial-utilizing-mitochondrial-illustrating-diuraphis-association-acyrthosiphon.webp)
+5
Related documents
