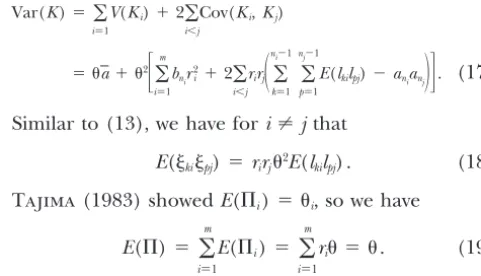Neutrality Tests Using DNA Polymorphism From Multiple Samples
Haipeng Li,*
,†Yunwu Zhang,
†Ya-Ping Zhang
†and Yun-Xin Fu*
,‡,1‡Laboratory of Bioinformatics, Yunnan University, Kunming 650991, People’s Republic of China,*Human Genetics Center,
University of Texas, Houston, Texas 77030 and†Laboratory of Molecular Evolution and Genome Diversity, Kunming
Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, People’s Republic of China
Manuscript received April 23, 2002 Accepted for publication November 21, 2002
ABSTRACT
The polymorphism of a gene or a locus is studied with increasing frequency by multiple laboratories or the same group at different times. Such practice results in polymorphism being revealed by different samples at different regions of the locus. Tests of neutrality have been widely conducted for polymorphism data but commonly used statistical tests cannot be applied directly to such data. This article provides a procedure to conduct a neutrality test and details are given for two commonly used tests. Applying the two new tests to the chemokine-receptor gene (CCR5) in humans, we found that the hypothesis that all mutations are selectively neutral cannot explain the observed pattern of DNA polymorphism.
STATISTICAL TESTS
T
HE amount and pattern of polymorphism in DNAsequence samples from a population reflects not A number of statistical tests can be extended for multi-only mutations in the ancestors of the sequences but ple samples. However, for the sake of discussion, we also random genetic drift as well as other evolutionary focus on two particular tests partly because they are forces, such as natural selection. How to detect the pres- used widely. First,Tajima (1989) proposed using the ence of natural selection in molecular population genet- difference between two estimates of (⫽4N), where ics and evolution is an important issue. It is possible to Nis the effective population size, andis the mutation detect the presence of natural selection because natural rate per sequence per generation, to detect the presence selection often causes the pattern of polymorphism to of selection. His test statistic is
differ from that under the neutral mutation hypothesis, which postulates that the majority of mutations that
DT⫽ 兿⫺
K/an
√
Var(兿⫺K/an), (1)
have contributed significantly to the genetic variation in natural populations is neutral or nearly neutral (Kimura
where⌸is the mean number of nucleotide differences 1983). However, the neutral mutation hypothesis is not
between two sequences,Kis the number of segregating sufficiently quantified to be tested rigorously in practice.
sites, which is equal to the total number of mutations A narrower definition of neutrality is that all mutations
under the infinite-sites model,nis sample size, and are selectively neutral, which is referred to as the
hypothe-sis of strict neutrality(FuandLi1993).
an⫽1⫹
1
2⫹. . . ⫹ 1
n⫺ 1. (2)
A number of statistical tests have been proposed, and almost all of them are designed for a single sample. In
reality, polymorphic data can be accumulated over time Second, Fu and Li (1993) suggested several tests of in the same or different laboratories, which means dif- neutrality, one of which is
ferent sites may be examined using different samples.
How to conduct neutrality tests in such situations is the D
F⫽
K⫺an1
√
Var(K⫺ an1), (3)
focus of this article. To date, millions of single nucleo-tide polymorphisms (SNPs) have been identified. It is
where 1 is the number of mutations in the external
very likely that SNPs from a single gene or tightly linked
branches, that is, mutations that are inherited by only regions will be typed for different samples by different
one sequence in the sample. research groups over time. The newly developed
The above two tests can be extended to multiple sam-method will be valuable for analyzing such data. We
ples in the following way. Assume that a locus without present an example of such an analysis using data from
recombination is divided intomregions that have been theCCR5 gene.
surveyed using different or partially overlapping sam-ples (Figure 1). It should be emphasized that the as-sumption of no recombination is made here to make the
1Corresponding author:Human Genetics Center, UT School of Public
null model as simple as possible, similar to the original Health, P.O. Box 20186, 1200 Herman Pressler, Houston, TX 77030.
E-mail: [email protected] Tajima and Fu and Li tests. Just as the presence of
⌸ ⫽
兺
mi⫽1
⌸i, (4)
K⫽
兺
m
i⫽1
Ki, (5)
and
1 ⫽
兺
m
i⫽1
1i. (6)
It is obvious that whenm⫽ 1, these equations reduce to their conventional definitions for a single sample. Note that in these equations the same weight is given to every region. An alternative approach is to give weight to a region according to certain criterion. However, so far we have not found any other weighting scheme to perform better than the equal-weighting scheme.
Figure1.—An illustration of a genealogy of multiple
sam-Furthermore, we define ples. The locus is divided into two regions. The sequence
length in the first region is 400 and in the second one is 600,
a ⫽
兺
m
i⫽1
riani, (7)
so r1⫽0.4 andr2⫽ 0.6 under the hypothesis of a constant
mutation rate. |S1|⫽3, |S2|⫽3, andS1傽S2⫽{3}. That means
{1, 2} are sequenced from 1 to 400, {4, 5} are sequenced from wherea
niis given by (2). That is,ais a weighted average
401 to 1000, and {3} is sequenced from 1 to 1000. The subtree
ofani. Then the tests (1) and (3) become
for the first region is marked by solid lines, and the subtree for the second region is labeled by dashed lines. The lengths
of branches can be calculated from coalescent times, and the D
T⫽
⌸ ⫺K/a
√
Var(⌸ ⫺K/a) (8) length of a branch here means a time duration. For example,g⫽t2⫹t3⫹t4, andh⫽t2. LetLibe the branch length of
the subtree for theith region, and thenL1⫽ a⫹b⫹g⫹ and
h⫹ e, andL2⫽ c⫹ d⫹ f⫹ e. The branch of eis shared
among two subtrees.lkiis the length of the k-size branch in D
F⫽
K⫺ a1
√
Var(K⫺a1). (9)
the subtree for theith region, sol11⫽a⫹b⫹h⫹e,l21⫽g,
l12⫽c⫹d⫹e, andl22⫽f. Five mutations are on the genealogy
and are shown as circles, three of which (solid circles) are found in the first region, and two of which (shaded circles)
are found in the second region. Therefore,11⫽2, 21⫽1, PERFORMING THE TESTS
12⫽2, and22⫽0.
Since Var(⌸ ⫺ K/a) ⫽ Var(⌸) ⫹ Var(K)/a2 ⫺ 2
Cov(⌸, K)/a and Var(K ⫺ a1) ⫽ Var(K) ⫹ a2 Var
(1)⫺2aCov(K,1), we can compute Var(⌸ ⫺K/a) and
recombination does not invalidate the Tajima and Fu Var(K⫺a1) if we are able to compute Var(K), Var(⌸),
and Li tests, but makes them more conservative, the Var(1), Cov(⌸,K), and Cov(K,1). Some of these terms
new tests will likely behave similarly and this will also can be computed analytically; others have to be
esti-be applicable to data with recombination. mated.
Define ⫽ 4N, where is the mutation rate per Analytical result:Assume the total sample consists of sequence per entire locus, andNis the effective popula- nsequences, and let those sequences be numbered from tion size. Also definei⫽4Ni, whereiis the mutation 1 ton.S⫽{1, 2, . . . ,n} will represent the whole sample.
rate for theith region (i⫽1, 2, . . . ,m), andri⫽ i/ The sample,Si, for theith region will be a subset ofS.
, which is the proportion of the mutation rate of the That is Si 債 S. We do not make any assumption here
ith region. If the mutation rate per site is constant, the on the relationship amongSi. We note that in one
ex-ri is equal to the proportion of the length of the ith treme, we can haveSi傽 Sj⫽ φfor every pair of iand
region, j, and on the other hand, we may haveS1⫽S2⫽. . .⫽
Sm ⫽ S. In many situations, it is likely thatSi 傽 Sj ⬆φ
兺
mi⫽1
ri ⫽1 . (e.g., Figure 1). We use |Si| to represent the number of
elements in the setSi, which is the sample size. Then
we haveni⫽ |Si|,n⫽|S|.
Furthermore, we define the mean number of nucleotide
differences between two sequences in theith region as The computation of Var(⌸), Var(K), and other com-ponents that we mentioned above requires
understand-⌸i, the number of segregating sites in theith region as
Ki, the number of mutations in the external branches ing of the sample genealogy. LetLibe the total branch
length of subtree for theith region scaled so that 1 unit of the genealogy in theith region as1i, and the sample
branch here means a time duration.kiis the number Var(K)⫽
兺
mi⫽1
V(Ki)⫹2
兺
i⬍jCov(Ki,Kj)
ofk-size mutations in theith region, andlkiis the length
ofk-size branches in the subtree for theith region scaled
⫽ a⫹ 2
冤
兺
m
i⫽1
bnir
2
i ⫹2
兺
i⬍jrirj
冢
兺
ni⫺1k⫽1
兺
nj⫺1
p⫽1
E(lkilpj)⫺anianj
冣冥
. (17)similarly as Li (Figure 1). The size of a branch is the
number of sequences in the sample that are descendants
Similar to (13), we have fori⬆jthat of that branch, and a mutation is said to besize kif it
occurs in a branch of sizek(Fu1995). Considering the E(kipj)⫽ rirj2E(l
kilpj) . (18)
subtree that is part of the tree shown as solid lines in
Tajima(1983) showedE(⌸i)⫽ i, so we have
Figure 1, the branch ofghas 2 descendent sequences, 1 and 2, so the size of the branch is 2. A mutation is
E(⌸)⫽
兺
m
i⫽1
E(⌸i)⫽
兺
m
i⫽1
ri ⫽ . (19)
on the branch of g, so the size of the mutation is 2. Following the definition of,i, andri, we have
Since⌸iis the mean number of nucleotide differences
i ⫽4Ni⫽ 4N(i/)⫽ri. (10) between two sequences in theith region, it can be
calcu-lated fromkias
Moreover, since 1 unit in time corresponds to 4N gener-ations, we have fromLiandFu(1998)
⌸i ⫽
2
ni(ni⫺1)
兺
ni⫺1
k⫽1
(ni⫺k)kki (20)
E(Li)⫽ani. (11)
(Fu1995).Tajima(1983) derived the variance of⌸ias
Under the infinite-sites model, we have
Var(⌸i)⫽
ni⫹1
3(ni⫺1)
ri ⫹
2(n2
i ⫹ni⫹ 3)
9ni(ni⫺ 1)
r2
i2. (21)
E(K)⫽
兺
m
i⫽1
E(Ki)⫽
兺
m
i⫽1
iE(Li)⫽ a. (12)
So we have Conditioning on the coalescent times, the number of
mutations in each branch follows a Poisson distribution E(⌸i⌸j)⫽E
冤
2
ni(ni⫺1) 兺
ni⫺1
k⫽1
(ni⫺k)kki
2
nj(nj⫺1) 兺
nj⫺1
p⫽1
(nj⫺p)ppj
冥
with parameterl, wherelis the branch length. We thus have
⫽ 42
ninj(ni⫺1)(nj⫺1)
冤
兺 ni⫺1k⫽1 兺
nj⫺1
p⫽1
kp(ni⫺k)(nj⫺p)rirjE(lkilpj)
冥
,E(KiKj|t2,t3, . . .)⫽ iLijLj ⫽rirj2LiLj, (22)
and thus wheretk(2ⱕkⱕ n) is the time duration required for
ksequences to coalesce tok⫺ 1 sequence,i.e., the
so-Var(⌸)⫽
兺
m
i⫽1
Var(⌸i)⫹2
兺
i⬍jE(⌸i⌸j) ⫺2
兺
i⬍j
[E(⌸i)E(⌸j)] ,
calledk-coalescent time (Figure 1), andi⬆j. Then we
have (23)
E(KiKj)⫽Et2,t3,…,tn[E(KiKj|t2,t3, . . . ,tn)]⫽rirj
2E(L
iLj) , where Var(⌸i) andE(⌸i⌸j) are given by (21) and (22),
(13) respectively. Moreover, from (12) and (19), we have
Cov(K,⌸)⫽E(K⌸)⫺E(K)E(⌸)⫽E(K⌸)⫺ 2a,
which leads to
(24) Cov(Ki,Kj)⫽E(KiKj)⫺E(Ki)E(Kj)⫽rirj2[E(LiLj)⫺anianj] .
where E(K⌸) is given later (Equation 25).Fu (1995) (14)
showed the formula to calculateE(kipi). After putting
these terms together, we have Watterson(1975) showed that Kin the case of one
sample is
E(K⌸)⫽E
冢
兺
m
i⫽1
Ki
兺
m
i⫽1 ⌸i
冣
⫽兺
m i,j
E
冤冢
兺
ni⫺1
k⫽1 ki
冣冢
2
nj(nj⫺1)
兺
nj⫺1
p⫽1
(nj⫺p)ppj
冣冥
Var(Ki)⫽anii⫹bni2i ⫽aniri ⫹bnir2i2, (15)
⫽
兺
mi⫽1
兺
ni⫺1
k⫽1
兺
ni⫺1
p⫽1
2(ni⫺p)p
ni(ni⫺1)
E(kipi)
where
⫹ 2
兺
i⬆j
兺
ni⫺1k⫽1
兺
nj⫺1
p⫽1
2(nj⫺p)p
nj(nj⫺1 )
rirjE(lkilpj) . (25)
bn⫽ 1⫹
1
4⫹ . . .⫹ 1 (n⫺ 1)2.
Also we can partitionE(LiLj) further as FromFu(1995), we have
E(LiLj)⫽ E
冢
兺
ni⫽1
k⫽1 lki
兺
nj⫺1
p⫽1 lpj
冣
兺
ni⫺1
k⫽1
兺
nj⫺1
p⫽1
E(lkilpj) . (16) E(ki)⫽
1
ki, (26)
TABLE 1
E(1)⫽
兺
m
i⫽1
E(1i)⫽
兺
m
i⫽1
i ⫽ (27)
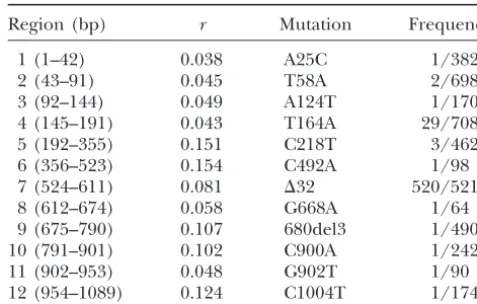
The list ofriand the frequencies of mutations in 12 regions of theCCR5 gene
and
Region (bp) r Mutation Frequency
Var(1)⫽Var
冢
兺
m
i⫽1
1i
冣
⫽兺
mi⫽1
Var(1i)
1 (1–42) 0.038 A25C 1/382
2 (43–91) 0.045 T58A 2/698
⫹2
兺
i⬍j
Cov(1i,1j). (28)
3 (92–144) 0.049 A124T 1/170
4 (145–191) 0.043 T164A 29/708
FuandLi(1993) showed that the variance of the total
5 (192–355) 0.151 C218T 3/462
number of mutations in the external branches is given 6 (356–523) 0.154 C492A 1/98
by 7 (524–611) 0.081 ⌬32 520/5210
8 (612–674) 0.058 G668A 1/64
Var(1i)⫽ i ⫹cni2i, (29)
9 (675–790) 0.107 680del3 1/490
10 (791–901) 0.102 C900A 1/242
wherecn⫽ 1 whenn ⫽2, and whenn⬎ 2
11 (902–953) 0.048 G902T 1/90
12 (954–1089) 0.124 C1004T 1/174
cn⫽2
nan⫺2(n⫺1)
(n⫺1)(n⫺ 2). CCR5 mutations and their frequencies in Caucasians come
from Carrington et al. (1997), and the frequency is the
From (18) and (26), we have
number of alleles observed/the total number of chromo-somes.
Cov(1i,1j)⫽E(1i1j)⫺E(1i)E(1j)
⫽rirj2E(l1il1j)⫺rirj2. (30)
Substituting (29) and (30) into (28), we have
the estimation can be improved by using large values ofM.
Var(1)⫽
兺
m
i⫽1
(ri ⫹cnir2i2)⫹2
兺
i⬍j(rirj2E(l1il1j)⫺rirj2),
The components mentioned above can be obtained (31) after E(lkilpj) is estimated, and then DT and DF can be
and calculated. Similar toTajima’s(1989) andFuandLi’s
(1993) tests,DT andDFdo not follow well-known
stan-Cov(K,1)⫽ E
冢
兺
m
i⫽1
Ki
兺
m
j⫽1
1j
冣
⫺ E(K)E(1) dard distributions. Since E(lkilpj) have to be estimatedfrom simulated samples, it is natural to use computer simulations to determine the critical points of the tests.
⫽
兺
mi⫽1
兺
ni⫺1
k⫽1
E(ki1i)⫹ 2
兺
i⬆j兺
ni⫺1
k⫽1
rirjE(lkil1j)⫺ 2a,
Overall, this approach gives more accurate critical values (32) than using approximation by a standard distribution.
whereE(ki1i) is given byFu(1995).
Numerical estimation: The above derivation shows AN EXAMPLE
that E(lkilpj) is critical for computing Var(K), Var(⌸),
We consider data from theCCR5 gene from Cauca-Cov(⌸,K), Var(1), and Cov(K,1). The value ofE(lkilpj)
sians (Carrington et al. 1997) to illustrate how the is dependent on the relationship of branches among
extended tests described in this article can be applied. subtrees of the regions. A simple example of the
rela-The CCR5 encodes a cell-surface chemokine-receptor tionship is given in Figure 1. Although Fu (1995) was
molecule that serves as a co-receptor for the macro-able to derive E(lkilpj) wheni ⫽ j, the general formula
phage-tropic strains of HIV-1. Because of its obvious fori⬆jappears to be analytically intractable. However,
importance, theCCR5 gene has been subjected to many an estimate can be obtained relatively easily from the
studies. One hypothesis is thatCCR5 might have been following procedure:
under natural selection (Carrington et al.1997). In 1. Simulate a genealogyg(topology without mutation) the data fromCarringtonet al.(1997), 12 mutations of a sample ofn sequences numbered from 1 ton. were documented in Caucasians, and 10 of the discov-2. Compute the value ofEg(lkilpj) for the simulated gene- ered mutations alter the amino acid sequence of the
alogy. protein, and each mutation is typed by different or
3. Repeat steps 1 and 2Mtimes. ThenE(lkilpj) is finally partially overlapping samples (Table 1). Since the
pre-estimated as cise relationships among samples were not given in the
original article, we consider two extreme cases here.
Eˆ(lkilpj)⫽
1
M
兺
gEg(lkilpj). In the first case, we assume that different samples are
composed of different individuals. That is,Si傽Sj⫽ φ,
wherei⬆ j. In the second case, we assume that smaller The computation ofEg(lkilpj) in step 2 is done in a similar
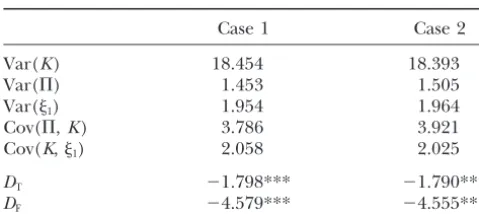
TABLE 2 cant negative values in both cases 1 and 2. Thus we conclude that the CCR5 region has not evolved
ac-Results of neutrality tests forCCR5
cording to the neutral model. One possibility is that it has evolved under natural selection, which remains to
Case 1 Case 2
be seen by further study.
Var(K) 18.454 18.393
We are grateful to Ms. Sara Barton for her help. This work is
Var(⌸) 1.453 1.505
supported by National Institutes of Health grants R01 GM50428 and
Var(1) 1.954 1.964
R01 GM55759 (Yun-Xin Fu), the Chinese Academy of Sciences
Cov(⌸,K) 3.786 3.921
(KSCX2-1-05), the National Nature Science Foundation of China, and
Cov(K,1) 2.058 2.025
the Nature Science Foundation of Yunnan Province in China.
DT ⫺1.798*** ⫺1.790***
DF ⫺4.579*** ⫺4.555***
LITERATURE CITED Statistical significance was calculated from the empirical
distribution ofDT andDF. ForDT, the critical values for the Carrington, M., T. Kissner, B. Gerrard, S. Ivanov, S. J. O’Brien
et al., 1997 Novel alleles of the chemokine-receptor geneCCR5.
1% significance tests are⫺1.663 (case 1) and⫺1.660 (case 2),
Am. J. Hum. Genet.61:1261–1267. and forDFthe values are⫺2.829 (case 1) and⫺2.631 (case 2).
Fu, Y. X., 1994 Estimating effective population-size or mutation-rate The values are estimated from 10,000 simulated samples.
using the frequencies of mutations of various classes in a sample ***The test result is significant at 1% level.
of DNA sequences. Genetics138:1375–1386.
Fu, Y. X., 1995 Statistical properties of segregating sites. Theor. Popul. Biol.48:172–197.
S8(64)傺S11(90)傺S6(98)傺S3(170)傺S12(174)傺S10 Fu, Y. X., andW. H. Li, 1993 Statistical tests of neutrality of
muta-tions. Genetics133:693–709. (242)傺S1(382)傺S5(462)傺S9(490)傺S2(698)傺S4
Kimura, M., 1983 The Neutral Theory of Molecular Evolution. Cam-(708)傺S7(5210), where the numbers in parentheses are bridge University Press, Cambridge, UK.
Li, W. H., andY. X. Fu, 1998 Coalescent theory and its applications the sample sizes. We refer to those two cases as case 1
in population genetics, pp. 45–79 inStatistics in Genetics, edited and case 2, respectively. By assuming samples are
inde-by E.Halloran. Springer-Verlag, New York.
pendent in the first case, we basically obtain the maxi- Tajima, F., 1983 Evolutionary relationship of DNA sequences in finite populations. Genetics105:437–460.
mum possible information from such data. On the other
Tajima, F., 1989 Statistical method for testing the neutral mutation hand, by assuming smaller samples are a subset of larger
hypothesis by DNA polymorphism. Genetics123:585–595. samples, we have the minimal amount of information. Watterson, G. A., 1975 On the number of segregating sites in
genetic models without recombination. Theor. Popul. Biol.7: From (12), we can get the estimate of as ˆ⫽K/a,
256–276. sincea⫽6.274, so we haveˆ⫽1.913. Also we have⌸ ⫽