Interallelic Complementation at the Mouse
Mitf
Locus
Eirı´kur Steingrı´msson,*
,1Heinz Arnheiter,
†Jo´n Hallsteinn Hallsson,*
,†M. Lynn Lamoreux,
2Neal G. Copeland
‡and Nancy A. Jenkins
‡*Department of Biochemistry and Molecular Biology, Faculty of Medicine, University of Iceland, 101 Reykjavı´k, Iceland, †Laboratory of Developmental Neurogenetics, National Institute of Neurological Disorders and Stroke, National
Institutes of Health, Bethesda, Maryland 20892 and‡Mouse Cancer Genetics Program, National Cancer Institute, NCI-FCRF, Frederick, Maryland 21702-1201
Manuscript received July 30, 2002 Accepted for publication October 15, 2002
ABSTRACT
Mutations at the mouse microphthalmia locus (Mitf) affect the development of different cell types, including melanocytes, retinal pigment epithelial cells of the eye, and osteoclasts. The MITF protein is a member of the MYC supergene family of basic-helix-loop-helix-leucine-zipper (bHLHZip) transcription factors and is known to regulate the expression of cell-specific target genes by binding DNA as homodimer or as heterodimer with related proteins. The many mutations isolated at the locus have different effects on the phenotype and can be arranged in an allelic series in which the phenotypes range from near normal to white microphthalmic animals with osteopetrosis. Previous investigations have shown that certain combinations ofMitfalleles complement each other, resulting in a phenotype more normal than that of each homozygote alone. Here we analyze this interallelic complementation in detail and show that it is limited to one particular allele,MitfMi-white(MitfMi-wh), a mutation affecting the DNA-binding domain. Both
loss- and gain-of-function mutations are complemented, as are otherMitfmutations affecting the DNA-binding domain. Furthermore, this behavior is not restricted to particular cell types: Both eye development and coat color phenotypes are complemented. Our analysis suggests thatMitfMi-wh-associated interallelic
complementation is due to the unique biochemical nature of this mutation.
M
UTATIONS at the mouse microphthalmia locus MitfMi-whmutation shows a severe heterozygouspheno-(Mitf) affect the development of several different type (large belly spots and coat color dilution) and an
cell types. Common to all the mutations are defects in intermediate homozygous phenotype (white coat and
the neural-crest-derived melanocytes, resulting in reduc- partial microphthalmia). Thus, the relative effects of
tion or lack of pigmentation in the coat, eye, and inner these two mutations are different in the hetero- and
ear. Most of the mutations also affect pigmented epithe- homozygous conditions. Although many moreMitf
mu-lial cells of the eye and mast cells, while only a few tations have been isolated since, Gru¨ neberg’s
observa-mutations result in osteoclast defects. Approximately tions still hold true; the semidominant phenotype of
one-half of theMitfalleles are semidominantly inherited theMitfMi-whmutation is the most severe phenotype
asso-and show white spotting asso-and/or coat color dilution ciated with any mutation at the locus while its
homozy-when heterozygous with wild type. The remaining alleles gous phenotype is only intermediate.Konyukhovand
are recessive and exhibit a phenotype only in homozy- Osipov(1968) analyzed the relationship between these
gous condition. TheMitfmi-spotted(Mitfmi-sp) mutation is
un-two alleles further and showed that heteroallelic animals
usual in that it displays a visible phenotype only in com- show interallelic complementation with respect to
ef-binations with other mutations at the locus (reviewed fects on the eye: MitfMi-wh/Mitfmi animals have normal
byMoore1995). eye size while homozygous littermates exhibit severe
In 1953, Gru¨ neberg noted the paradoxical relations (Mitfmi/Mitfmi) or intermediate (MitfMi-wh/MitfMi-wh)
mi-of the semidominantMitfmicrophthalmia(Mitfmi) andMitfMi-wh
crophthalmia. Similar analysis with a few otherMitf
al-mutations (Gru¨ neberg 1953). While theMitfmi
muta-leles also showed interallelic complementation. For
ex-tion exhibits a weak phenotype in the heterozygous ample, Schaible (1963) showed that a combination
condition (occasional head blaze and belly streaks) and of theMitfMi-wh allele with the Mitfmi-black-eyed white (Mitfmi-bw)
a very strong phenotype in the homozygous condition mutation resulted in white animals with pale yellow spots
(microphthalmia, white coat, and osteopetrosis), the (on the C3HxB6 background they have pigmented spots
on the back). Homozygotes for both mutations are com-pletely white. Similarly, heteroallelic combinations of
1Corresponding author:Department of Biochemistry and Molecular
MitfMi-wh with Mitfmi-white spot (Mitfmi-ws) produced animals Biology, Faculty of Medicine, University of Iceland, Vatnsmy´rarvegur
with dark eyes of normal size and a spotted, checker-16, 101 Reykjavı´k, Iceland. E-mail: [email protected]
2Present address:8255 Sandy Point Road, Bryan, TX 77807. board-like coat with yellowish-brown to gray colors
MATERIALS AND METHODS
(Hollander1964). Homozygotes for theMitf
muta-tion are white with pink eyes. This suggests that interal- The followingMitfmutants were used in this study: C57BL/
lelic complementation is a common phenomenon at 6J-MitfMi-wh, C57BL/6J-Mitfmi-sp, C57BL/6J-Mitfmi-red-eyed white(Mitfmi-rw),
C57BL/6J-Mitfmi-eyeless white(Mitfmi-ew), C57BL/10-Mitfmi-black and white spot
theMitflocus and that both eye and coat color defects
(Mitfmi-bws), C57BL/6J-Mitfmi, 82UT-MitfMi-oak ridge (MitfMi-or),
82UT-are affected. Although this phenomenon has never been
Mitfmi-brownish (Mitfmi-b), and mixed [C3H/C57BL/6J]-Mitfmi-vga-9
characterized systematically in detail, these observations
(Table 1). These strains are maintained at the National Cancer
suggest that interallelic complementation reveals an im- Institute in Frederick, Maryland, and at the National Institute
portant aspect of the nature of theMitflocus. of Neurological Disorders and Stroke, National Institutes of
Health, in Bethesda, Maryland. The mice were mated
systemat-The MITF protein is a member of the MYC supergene
ically to generate the different allelic combinations. At least
family of basic-helix-loop-helix-leucine zipper
(bHLH-three different independent crosses were set up for generating
Zip) transcription factors and is most closely related to each combination and multiple offspring (⬎25) were analyzed
the TFE3, TFEB, and TFEC proteins (Hodgkinson et from each cross. The phenotypes of the resulting animals were
al. 1993; Hugheset al. 1993). Like other members of visually inspected, the animals were photographed at 6 weeks
of age (or earlier), and eyes, skin, and Harderian gland were
the bHLH-Zip family, MITF has been shown to bind
dissected for histologic analysis. All tissue specimens were fixed
the CANNTG E-box sequencein vitroas either a
homo-in Bouhomo-ins fixative, sectioned, and then stahomo-ined with
hemotoxy-dimer or a heterohemotoxy-dimer with TFE3, TFEC, and TFEB lin and eosin. For the evaluation of melanin in Harderian
(Hemesathet al. 1994). The basic domain is the DNA- glands and in the retinal pigment epithelium (RPE) of the
binding domain of the protein while the HLH and Zip eye, tissues were stained with Fontana-Masson.
domains are responsible for dimerization. Consistent with its role as a regulator of gene expression, MITF is
RESULTS
primarily located in the nucleus (Takebayashi et al.
1996) where it can activate expression from pigment Heteroallelic combinations ofMitfmutations:To
per-cell, mast per-cell, and osteoclast specific promoters (Bent- form a systematic study of theMitf-associated interallelic
leyet al. 1994;Yasumotoet al. 1994;Moriiet al. 1996; complementation, availableMitfalleles were crossed to
YasumotoandShibahara1997;Motyckovaet al. 2001). each other in all possible combinations. The phenotypes
The molecular and biochemical defects associated of the resulting heteroallelic offspring were studied by
with most of the Mitf alleles have been determined visual inspection of coat color and eye size. The alleles
(Hodgkinsonet al. 1993;Hugheset al. 1993;Hemesath used in this study are described in Table 1 and range in
et al. 1994; Steingrı´msson et al. 1994, 1996; Yajima phenotype from very mild (e.g.,Mitfmi-sp) to severe (
Mitfmi)
et al. 1999; Hallssonet al. 2000). The semidominant and in mode of inheritance from recessive (Mitfmi-sp,
mutations characterized to date affect either the DNA- Mitfmi-rw,Mitfmi-ew, Mitfmi-vga9) to semidominant (MitfMi-wh,
binding or the transcriptional activation domains of the MitfMi-or, MitfMi-b, Mitfmi). In all the crosses made, the
protein while dimerization domains are unaffected number of heteroallelic progeny was according to
Men-(Hodgkinson et al. 1993; Steingrı´msson et al. 1994, delian ratios (data not shown) and the phenotypes were
1996). The mutant proteins cannot bind DNA; however, consistent among the different progeny and litters of
they can still dimerize with proteins such as TFE3 and each cross.
thereby interfere with DNA binding of the partner The phenotypes of the resulting combinations are
(Hemesathet al. 1994;Steingrı´mssonet al. 1996). The described in Table 2 and in Figures 1–3. In most cases
dominant-negative behavior of these mutant proteins where the homozygous phenotypes of the two alleles
in vitrothus accounts for the phenotype seen in heterozy- are similar to each other, the phenotypes of the
hetero-gous mice. Consistent with this, the recessive mutations allelic combinations are the same as each of the
homozy-affect either the dimerization domain of the MITF pro- gotes. For example, MitfMi-or/Mitfmi animals are white
tein or the transcription of theMitfgene, resulting in and microphthalmic and have severe osteopetrosis just
little or no MITF production (Hodgkinsonet al. 1993; like the respective homozygotes (MitfMi-or/MitfMi-or and
Hugheset al. 1993;Steingrı´mssonet al. 1994;Yajima Mitfmi/Mitfmi; Table 2). Similarly,Mitfmi-ew/Mitfmi-vga9
ani-et al. 1999). mals have a white coat and severe microphthalmia but no
Despite the detailed molecular and biochemical anal- osteopetrosis, just like theMitfmi-ew/Mitfmi-ewandMitfmi-vga9/
ysis of the Mitfmutations, no satisfactory explanation Mitfmi-vga9homozygotes (Table 2). In most cases in which
for the interallelic complementation has emerged. Here, the homozygous phenotypes are different, however, the
we perform a detailed genetic analysis of the interallelic heteroallelic animals exhibit an intermediate
pheno-complementation at the Mitf locus and show that it type. For example,Mitfmi-ew/
Mitfmi-spanimals have white
is restricted to the MitfMi-whmutation, a mutation with feet, head, and belly while the rest of the coat is gray
unique characteristics. Our analysis suggests that the (Table 2 and Figure 1A). This phenotype is intermediate
nature of theMitf-associated interallelic complementation between the white microphthalmicMitfmi-ew/Mitfmi-ew
(Ta-ble 2) and near normalMitfmi-sp/Mitfmi-sp(Table 2, Figure
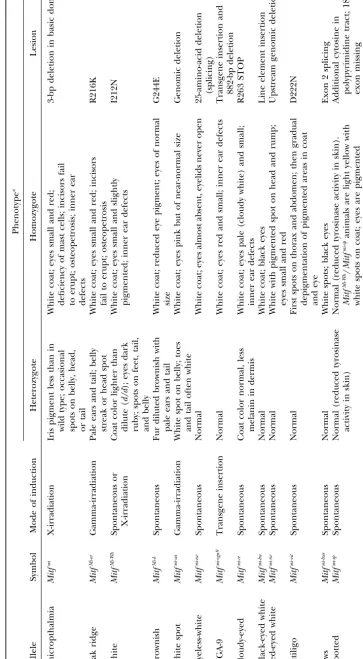
Figure1.—Phenotypes resulting fromMitf mutations and heteroallelic combinations. (A)Mitfmi-sp/Mitfmi-ew. (B)Mitfmi-sp/Mitfmi-sp:
Note the apparently normal appearance of this animal. (C)MitfMi-b/Mitfmi-rw: The pigmented spots are agouti due to the C3H
background of the MitfMi-b mutation. (D) Mitfmi-rw/Mitfmi-rw: Note the black head spots. (E) Mitfmi-ew/Mitfmi: This animal lacks
pigment and shows severe microphthalmia but no osteopetrosis. (F) MitfMi-wh/Mitfmi-sp. (G) Mitfmi-rw/Mitfmi: Note the severe
microphthalmia. (H)MitfMi-wh/Mitfmi-rw: Note the faintly pigmented patch on their heads (dotted line).
1B) animals. Similarly,MitfMi-b/Mitfmi-rwanimals are white white spots and normal-size eyes (WolfeandColeman
1964; Table 2, Figure 1F). Although Mitfmi-sp
homozy-with large pigmented spots (Table 2 and Figure 1C),
a phenotype intermediate between the white MitfMi-b gotes have a normal appearance, a reduction in the
activity of the tyrosinase enzyme has been detected in
homozygotes (Table 2) and Mitfmi-rw/Mitfmi-rw animals,
which are white with a pigmented spot on the head their skin (WolfeandColeman1964). All heteroallelic
combinations involvingMitfmi-spshow a more severe
de-(Table 2, Figure 1D). In all these cases, one of the alleles
encodes a partially functional protein. Interestingly, the fect in pigmentation than these mutations show in
het-erozygous combinations with wild type, albeit less than
combination of Mitfmi-ew andMitfmiresults in white,
se-verely microphthalmic animals (Figure 1E); this pheno- what is observed in homozygotes for the other allele
(Table 2). For example,Mitfmi-rw/Mitfmi-spanimals have
type is identical to that ofMitfmi-ew homozygotes while
Mitfmihomozygous animals also exhibit osteopetrosis. a large belly spot (Table 2), a phenotype not seen in
either Mitfmi-rw/⫹ or Mitfmi-sp/Mitfmi-sp animals. Also,
All combinations involving the Mitfmi-spmutation fall
into the intermediate group; the resulting phenotypes MitfMi-b/Mitfmi-spanimals exhibit a diluted coat color
phe-notype while MitfMi-or/Mitfmi-sp animals are white with
are in direct relation to the severity of the allele to which
Mitfmi-spis crossed (Table 2). This mutation was originally spots that gradually lose their pigmentation with age
(Table 2). This gradual depigmentation is similar to
found in a colony ofMitfMi-whanimals (Wolfeand
Cole-man1964). While animals homozygous for theMitfmi-sp that observed for the Mitfmi-vitilago (Mitfmi-vit) mutation
(Lerneret al. 1986;Lamoreuxet al. 1992). mutation cannot be distinguished from their wild-type
littermates,MitfMi-wh/Mitfmi-spheterozygotes are tan with It is interesting to compare the phenotypes ofMitfmi-sp
Figure2.—Phenotypes asso-ciated with combinations with the Mitfmi-vga9
loss-of-func-tion mutaloss-of-func-tion. (A) Mitfmi-vga9/ Mitfmi-vga9: Note the severe
mi-crophthalmia. (B) Mitfmi-ew/ Mitfmi-vga9. (C)MitfMi-wh/Mitfmi-vga9:
Figure3.—Interallelic complementation at theMitflocus. (A)MitfMi-wh/⫹(pigmented, in the back) andMitfMi-wh/MitfMi-wh(white
and microphthalmic, in the front) animals: Note the intermediate microphthalmia. (B)Mitfmi/Mitfmi: Note the severe microphthalmia
and small size of the 3-week-old animals. (C)MitfMi-wh/Mitfmi. Eyes are of normal size (arrows) although eye pigment is somewhat
reduced compared to normal. (D)MitfMi-wh/Mitfmi-ew. The eyes are of normal size (arrows). (E)MitfMi-or/MitfMi-or(front) andMitfMi-wh/ MitfMi-or(back) animals. Note the normal eye size in the compound heterozygote (arrow). (F)MitfMi-wh/MitfMi-bandMitfMi-b/MitfMi-b
animals. The two compound heterozygotes (leftmost animal and the animal on top) show light-brown coat color while the two MitfMi-bhomozygotes (bottom) are completely white. Neither genotype affects eye size. (G) Mitfmi-bws/Mitfmi-bws: Note the black
spots on otherwise white background. Eye size is normal. (H)MitfMi-wh/Mitfmi-bws. The phenotype is intermediate between each
homozygote (compare to A and G) and no complementation is observed.
homo- and heterozygotes with the phenotypes of het- supported by genetic studies (Steingrı´msson et al.
2002; see below). eroallelic combinations involving the loss-of-function
Mitfmi--vga9and the recessiveMitfmi-ewmutations. As already The Mitfmi-rw mutation also has unique features.
Ho-mozygotes for this mutation are white with pigmented
explained, no phenotype is visible in Mitfmi-sp/
Mitfmi-sp
animals, suggesting that the mutant protein has some spots of somewhat variable size on the head (Figure
1D); occasional pigmented spots are observed in other
activity. However, one dose of the Mitfmi-sp mutant
pro-tein is clearly not sufficient for proper melanocyte de- body regions, including the rump. Although the eyes
are smaller than normal, appear red, and lack pigment velopment since heteroallelic combinations involving
Mitfmi-vga9 and Mitfmi-ew result in defective pigmentation altogether, they are not as severely affected as animals
carrying the loss-of-function Mitfmi-vga9 mutation or in
(Table 2, Figures 1 and 2). Interestingly, although the
homozygous phenotypes of the Mitfmi-vga9 and Mitfmi-ew MitfMi-whanimals (Table 2; Figure 3A). TheMitfmi-rw
muta-tion is caused by the lack of a large pormuta-tion of theMitf
mutations are almost identical, the heteroallelic
combi-nations differ in that Mitfmi-vga9/Mitfmi-sp animals have regulatory region, resulting in the absence of the 5⬘
exons 1h and 1b and in aberrant expression of the gene
large pigmented spots while Mitfmi-ew/
Mitfmi-sp animals
have a uniformly gray coat color over most of their body; (Steingrı´mssonet al. 1994;Hallssonet al. 2000). Most
heteroallelic combinations involving Mitfmi-rw result in
head and feet are white. This reflects the fact that the
Mitfmi-vga9 mutation is a loss-of-function mutation; the white microphthalmic animals: Combinations with the
Mitfmi,
MitfMi-or, and
Mitfmi-ewmutations all produce white
heteroallelic combination reveals the function of a
sin-gle copy of the Mitfmi-sp protein. The Mitfmi-ew/Mitfmi-sp microphthalmic animals (Table 2; Figure 1G) and
Mitfmi-rw/Mitfmi-vga9animals are white and microphthalmic
combination, on the other hand, reveals the partial
dominant-negative nature of the Mitfmi-ew protein. Al- and most have a small pigmented head spot (Table 2;
Figure 2). In all these cases the microphthalmia
ob-though theMitfmi-ewmutation behaves in a recessive
fash-ion on its own and is classified as such in this study,in served is more severe than that inMitfmi-rw
homozygotes.
However, combinations of the Mitfmi-rw mutation with
vitrostudies suggest that the mutant protein has strong
dominant-negative activity (Hemesath et al. 1994). MitfMi-bandMitfMi-whresult in somewhat milder
pheno-types: Mitfmi-rw/MitfMi-b animals are white with an
occa-However, the Mitfmi-ew protein is unable to enter the
nucleus efficiently (Takebayashiet al. 1996;Nakayama sional pigmented spot and normal eye size (Table 2) while
Mitfmi-rw/MitfMi-whanimals are white with a tan head spot;
et al. 1998), resulting in a dominant-negative
cyto-plasmic protein. Since the protein needs to enter the eye size is near normal and lacks pigment (Figure 1H).
In all the cases described above, coat color is most nucleus to realize its effects, this may result in only
minor dominant-negative effectsin vivo, at least in some sensitive to mutations atMitf, indicating that
melano-cytes have the highest requirement for Mitf function.
Eye color follows, then eye size, and only a few of the notypes in homozygous condition, the third (Mitfmi-rw) is
more severely affected; all three have milder phenotypes mutations show osteopetrosis. Thus, the pigment cells
of the retinal epithelium of the eye have a lower require- than that of MitfMi-wh in homozygous condition. Both
semidominant and recessive mutations are
comple-ment forMitfprotein than do melanocytes, and
osteo-clasts seem to be able to function with very low amounts mented while only recessive mutations fail to be
comple-(if any) ofMitfin the cell. mented (Tables 1 and 2).
Interallelic complementation at Mitf: In contrast to This is further supported by previously reported
het-the results described above, most of het-the combinations eroallelic combinations involvingMitfMi-wh. For example,
involving the MitfMi-wh allele show interallelic
comple-Mitfmi-bw/
MitfMi-whare white with some pale yellow spots,
mentation in which the resulting phenotype is more which depigment with age, whereas homozygotes for
normal than that of each of the homozygotes alone. This the recessiveMitfmi-bwmutation are white with black eyes
is true for combinations involving the semidominant (Schaible 1963). The Mitfmi-ws mutation is
semidomi-mutations Mitfmi,
MitfMi-or, and
MitfMi-b, as well as the
nant; heterozygotes have a white belly spot while
homo-recessiveMitfmi-ewmutation and the
Mitfmi-vga9
loss-of-func-zygotes are white, most with normal eye size.
Heteroal-tion mutaHeteroal-tion. All these combinaHeteroal-tions, exceptMitfMi-wh/
lelic combinations of this mutation withMitfMi-whresult
MitfMi-b, result in white animals with normal eye size
in spotted animals with yellowish-brown-to-gray spots
(Table 2; Figure 3, A–E). The normal eye size is in sharp that depigment with age (Hollander1968). Thus, coat
contrast to the severe microphthalmia observed in each color is complemented. TheMitfmi-diandMitfmi-ce
muta-of the MitfMi-wh, Mitfmi, MitfMi-or, Mitfmi-ew, and Mitfmi-vga9
tions have very similar phenotypes in homozygous
con-homozygotes (Table 2; Figures 2 and 3). The heteroal- dition (white coat and small eyes) and, in fact, the
mo-lelic combination ofMitfMi-whandMitfMi-bresults in
light-lecular defect involved is identical (Hallsson et al.
tan animals with occasional white spots (Figure 3F); 2000). Consistent with this, heterozygous combinations
while eye size is normal, the RPE layer contains some ofMitfmi-diandMitfmi-cewithMitfMi-whresult in normal eye
pigment (data not shown). Although the eye size of this size, showing that both mutations are complemented
heteroallelic combination is more normal than that of (Westet al. 1985; M. L.Lamoreux, unpublished
obser-MitfMi-whhomozygotes,MitfMi-b/MitfMi-banimals have
nor-vations). mal eye size. Thus, the eye size of the heteroallelic
com-bination is similar to that of one of the homozygotes so
no complementation is observed with respect to this DISCUSSION
feature. However, coat color pigmentation is more
nor-The allelic series at the Mitf locus shows that the
mal in the heteroallelic combination than in each of the
three major cell types affected by mutations at the locus, homozygotes, suggesting complementation with respect
melanocytes, osteoclasts, and retinal pigment epithelial
to this phenotype. Clearly, the MitfMi-wh mutation can
cells of the eye, have different requirements for the Mitf complement both the microphthalmia and the coat
protein: Most of theMitfmutations affect pigmentation
color phenotypes of the differentMitfmutations.
of the coat, many affect eye development, and only a Interestingly, three recessive mutations are not
com-few result in osteopetrosis. Clearly, the requirement for
plemented byMitfMi-wh (Table 2). As explained above,
Mitffunction is very different in the cell types in which
the heteroallelic combination MitfMi-wh/Mitfmi-sp results
the highest requirement is observed in melanocytes and in tan animals with occasional white spot. This
pheno-the lowest in osteoclasts. Osteopetrosis is observed only type is intermediate between the two homozygotes and
in homozygotes for theMitfmiandMitfMi-ormutations as
therefore no complementation is observed (Table 2).
well as in compound heterozygotes of these mutations
MitfMi-wh/Mitfmi-rwanimals also have an intermediate
phe-(Table 2). No osteopetrosis is observed inMitfMi-wh
ho-notype although some complementation may be
ob-mozygotes or in any of the other mutations tested, ex-served with respect to eye development. In these animals
ceptMitfmi-ew, which shows mild hyperosteosis in the
ho-coat color is white with tan head spots while eye size
mozygous condition (Steingrı´msson et al. 2002).
varies from microphthalmic to near normal (Figure
Although theMitfmi-ewmutation is classified as recessive
1H); the difference in eye size can be bilateral with
in this study, the mutant protein has partial dominant-one eye near normal and the other microphthalmic.
negative activity, at least in osteoclasts. Recent studies
Perhaps most interestingly, the MitfMi-wh/Mitfmi-bws
com-have shown that although osteoclasts are normal inMitf
pound heterozygotes have an intermediate phenotype
null mice as well as in mice carrying a loss-of-function showing tan spots and normal eye size (Figure 3H). In
mutation in the Mitf-related gene Tfe3, the combined
the homozygous condition theMitfmi-bwsmutation results
loss of both genes results in severe osteopetrosis (
Stein-in large, heavily pigmented spots (Table 1, Figure 3G)
grı´msson et al. 2002). Thus, the Mitf-associated os-and normal eye size. Thus, the phenotype of the
hetero-teopetrosis is seen only upon simultaneous loss of Mitf allelic combination is not complemented. While two of
and Tfe3 activity in osteoclasts. Only dominant-negative the recessive mutations that fail to be complemented
show this phenotype. The osteoclast phenotype never fail to be complemented byMitf are mutations that affect regulatory regions as well as mutations that affect shows interallelic complementation.
The phenotype ofMitfMi-whmutant animals is unusual protein-coding regions. The common theme among the
mutations that fail to be complemented is that they have in that heterozygotes are severely affected while
homozy-gotes have an intermediate phenotype compared to that milder effects in the homozygous condition thanMitfMi-wh
and the mutations that are complemented byMitfMi-wh.
of the other Mitf alleles: In heterozygotes coat color
is severely affected while eye development is not; in This does not fit the criteria of transvection.
Despite the fact that the MITF protein can form both homozygotes, coat color is still severely affected while
eye development has an intermediate phenotype. This homo- and heterodimers, it is difficult to see how the
Mitf-associated complementation could be due to the
suggests different effects of the mutation in the two
pigment cells affected, melanocytes and RPE cells. Fur- formation of functional dimers by two different mutant
proteins. Three of the mutations that show interallelic thermore, it suggests that the dominant-negative nature
of theMitfMi-whmutation has much more serious effects complementation with
MitfMi-wh (
Mitfmi-ew,
Mitfmi-mi, and MitfMi-or) all affect the DNA-binding basic domain, the
in melanocytes than in RPE cells.
In the eye, complementation is observed in many very same domain affected by theMitfMi-whmutation
(Ta-ble 1;Steingrı´mssonet al. 1994). The fourth mutation
different combinations withMitfMi-wh, even inMitfMi-wh/
Mitfmi-vga9 animals in which the MitfMi-wh allele is paired to show interallelic complementation isMitfmi-b, whose
molecular defect is in the loop of the HLH domain and with a loss-of-function mutation. Coat color is
comple-mented only whenMitfMi-whis paired with hypomorphic also affects DNA-binding abilities of the protein (Table
1;Steingrı´mssonet al. 1996). Thus, all these mutations
mutations such as MitfMi-b and Mitfmi-ws, which already
have normal eye development (Hollander1964). This affect DNA binding. Most interestingly, the
loss-of-func-tion mutaloss-of-func-tion Mitfmi-vga9 can complement the MitfMi-wh
suggests that the complementation occurs in a reverse
allelic series, again reflecting the activity requirement mutation, suggesting that for complementation to take
place it is better to have no or very little Mitf activity
for Mitf in the different cell types. The threshold for
theMitfrequirement is lower in RPE cells than in mela- with theMitfMi-whmutation than to have partial function.
These facts are difficult to reconcile with the model of nocytes and therefore the eye phenotype is more easily
complemented than the coat color phenotype. Interest- complementation by active dimers.
Our observations lead us to propose an alternative ingly, no complementation is observed in combinations
with alleles with a phenotype more normal than that of dose-dependent model of interallelic complementation
(Figure 4). According to this model, the phenotype
MitfMi-whin homozygous condition.
To date, two main models have been proposed to is determined by the level of the MitfMi-wh protein in
combination with a cell-type-specific level of sensitivity explain interallelic complementation. The first model
involves transvection in which one allele affects the ex- to this protein. Earlier studies have shown that like
wild-type Mitf, the MitfMi-wh protein comes in two distinct
pression of a second allele on the homologous
chromo-some. Generally, these involve combinations of a regula- isoforms, which differ in the presence or absence of six
residues upstream of the basic domain (Steingrı´msson
tory mutation with a mutation in the coding region.
Transvection depends on chromosomal pairing and et al. 1994). The alternative six amino acids are the result
of alternative splice acceptor sites in exon 6; all tissues chromosomal rearrangements have been shown to
in-terfere with the complementation. The best examples analyzed so far contain both splice forms (Hallsson
et al. 2000). In vitro, the MitfMi-wh protein lacking the
of this involve theUltrabithorax(Ubx) gene in Drosophila
where pairing of certain alleles results in partial comple- alternative six amino acids acts in a dominant-negative
fashion; the protein can dimerize with partner proteins
mentation of the phenotype (reviewed by Pirrotta
1999). The second model for interallelic complementa- but cannot bind DNA and interferes with the DNA
bind-ing of the related Tfe3 protein in vitro (Hemesath et
tion involves dimer formation between protein
mono-mers expressed by two different alleles. The different al. 1994). However, although the MitfMi-whprotein
con-taining the alternative six amino acids cannot bind DNA alleles affect different functional domains and the
re-sulting dimer is therefore partially functional and com- as a homodimer, it can bind DNA as a heterodimer
with Tfe3 at levels similar to the wild-type Mitf protein plementation is observed. Examples of this include the
Egfrlocus in Drosophila (Razet al. 1991) and thelet-23 (Hemesathet al. 1994). Thus, the mutation produces
two different proteins with critically altered function: a
gene inCaenorhabditis elegans(Aroianet al. 1994).
In the case ofMitfreported here, neither of these two dominant-negative protein [MitfMi-wh(⫺6)] and a
pro-tein that cannot bind DNA as a homodimer, yet can models clearly apply. Transvection is a highly unlikely
explanation forMitf-associated interallelic complemen- bind DNA as a heterodimer [MitfMi-wh(⫹6)] (Figure 4A)
with its partners. Due to the intermediate phenotype of tation. The only allele that results in complementation
(MitfMi-wh) does not detectably affect regulation of the MitfMi-wh homozygous animals, one of these proteins
must be partly functional. gene and is a mutation in the protein-coding region
Figure4.—The biochemi-cal behavior of the MitfMi-wh protein provides a model for the interallelic complementa-tion at Mitf. (A) The bio-chemical behavior of the two different MitfMi-wh proteins. (B) A model depicting the ef-fects of the mutant proteins on the phenotype. Yellow bars represent levels of nor-mal Mitf function while red bars indicate levels of neo-morphic activity. The dotted lines indicate threshold re-quirements for each type of activity. The thresholds can be different in the different cell types affected. Three dif-ferent situations are shown: wild type, MitfMi-wh
homo-zygotes, and the MitfMi-wh/ Mitfmi-vga9heteroallelic
combi-nation. In wild type (left), two different messages are expressed in all tissues analyzed, leading to the synthesis of two different Mitf proteins: The Mitf(⫺6) protein has 20% less stability in a complex with DNA than the Mitf(⫹6) protein (Hemesathet al. 1994). InMitfMi-whhomozygotes
(middle), the MitfMi-wh(⫹6) protein is more or less normal as a heterodimer while the MitfMi-wh(⫺6) protein cannot bind DNA as a homo- or heterodimer. In addition, it has acquired new (neomorphic) properties, which negatively affect the phenotype. The resulting phenotype is a trade-off between the two different effects and differs in cell types. In the MitfMi-wh/Mitfmi-vga9
heteroallelic combination (right) the dose of neomorphic activity has been reduced by one-half such that it is below the threshold required to see effects on the phenotype. At the same time, the normal activity of the MitfMi-wh(⫹6) protein is sufficient to allow normal function and complementation is observed.
protein is likely to provide the explanation for both From the mutant phenotype it is clear that the effects
of theMitfMi-whmutation are relatively more serious in
the interallelic complementation associated with this
mutation and the fact thatMitfMi-whhas the most severe the neural-crest-derived melanocytes than in RPE cells
or osteoclasts. Coat color is severely affected inMitfMi-wh
phenotype in heterozygous condition. We therefore
propose that the genetic behavior of this mutation is homo- and heterozygotes while only homozygotes show
intermediate microphthalmia; bone development is due to the relative effects of homo- and heterodimers
involving this mutation where the MitfMi-wh(⫺6) protein normal in both cases. This suggests a tissue-specific
dif-ference in the effects of theMitfMi-whmutation. Perhaps
is very efficient as a dominant-negative protein and
where MitfMi-wh(⫹6) is at least partly functional. The two this is an indication that the mutant protein interacts
with one or more melanocyte-specific proteins. Thus,
different versions of the MitfMi-wh protein may not be
able to form DNA-binding dimers between themselves in MitfMi-wh/⫹ animals, the strong dominant-negative
action of the MitfMi-wh(⫺6) protein may interfere with
but may be able to dimerize with other partner proteins
in the cell. Furthermore, we propose that one of the two partner proteins in melanocytes. Alternatively, the new
(neomorphic) activity may negatively affect the function
isoforms of the MitfMi-whprotein (or both) has acquired a
new (neomorphic) function, resulting in negative ef- of melanocyte-specific factors or processes. Together,
these effects result in the severe coat color phenotype fects in the cell. This new action may be the result of
dominant-negative action of the MitfMi-wh(⫺6) protein observed in heterozygotes. Although the RPE cells of
MitfMi-wh/⫹ and
MitfMi-wh/
MitfMi-wh animals also contain
against essential proteins or pathways in the cell. It may
also be due to a novel action of the MitfMi-wh(⫹6) protein, the neomorphic activity (since they also express the
mutantMitfgene), RPE cells are not as severely affected
e.g., binding the wrong promoter sequence or activating
the wrong set of genes with subsequent negative effects. since the activity threshold is different (Figure 4B) and
the RPE cells do not express the melanocyte-specific For the following discussion we assume that the new
(neomorphic) activity is associated with the MitfMi-wh protein(s) against which the dominant-negative MitfMi-wh
(⫺6) protein acts.
(⫺6) protein. Finally, we propose that the different
activities of the two forms of the MitfMi-whprotein each The intermediate eye phenotype observed in MitfMi-wh
homozygotes supports the idea that the MitfMi-wh(⫹6)
have their own threshold requirements in the different
cell types affected. A model depicting theMitf-associated protein has partially normal function in RPE cells. The
partially normal function of the MitfMi-wh(⫹6) protein
is unaffected by the dominant-negative activity of the
at themilocus. J. Biol. Chem.268:20687–20690. MitfMi-wh(⫺6) protein since MitfMi-wh(⫹6) is unable to form
Konyukhov, B. V., andV. V. Osipov, 1968 Interallelic
complemen-tation of microphthalmia and white genes in mice. Genetika4:
DNA-binding homodimers with other mutant MitfMi-wh
65–76.
proteins (Hemesathet al.1994). In the different
hetero-Lamoreux, M. L., R. E. Boissy, J. E. WomackandJ. J. Nordlund,
allelic combinations, the new (neomorphic) action of 1992 Thevitgene maps to themi(Micropthalmia) locus of the
laboratory mouse. J. Hered.83:435–439.
MitfMi-whhas been reduced by half as compared to that
Lerner, A. B., T. Shiohara, R. E. Boissy, K. A. Jacobson, M. L. of MitfMi-wh homozygotes, thereby reducing the effects
Lamoreuxet al., 1986 A mouse model for vitiligo. J. Invest.
of neomorphic activity below a certain threshold level Dermatol.87:299–304.
Moore, K. J., 1995 Insight into the microphthalmia gene. Trends
and allowing more normal development to proceed
Genet.11:442–448. (Figure 4B). Thus, the heteroallelic combination of
Morii, E., T. Tsujimura, T. Jippo, K. Hashimoto, K. Takebayashi
MitfMi-wh and Mitfmi-vga9 uncovers the partially normal
et al., 1996 Regulation of mouse mast cell protease 6 gene ex-pression by transcription factor encoded by themilocus. Blood
function of the MitfMi-wh(⫹6) protein, which is sufficient
88:2488–2494. to generate eyes of normal size. In heteroallelic
combi-Motyckova, G., K. N. Weilbaecher, M. Horstmann, D. J. Rieman,
nations with milderMitfmutations that still retain some D. Z. Fisheret al., 2001 Linking osteopetrosis and
pycnodys-ostosis: regulation of cathepsin K expression by the
microphthal-Mitf activity, the dominant-negative MitfMi-wh(⫺6)
pro-mia transcription factor family. Proc. Natl. Acad. Sci. USA98:
tein interferes with the function of the mutant proteins
5798–5803.
and an intermediate phenotype is observed. The neo- Nakayama, A., M.-T. T. Nguyen, C. C. Chen, K. Opdecamp, C. A.
Hodgkinsonet al., 1998 Mutations inmicrophthalmia, the mouse
morphic activity is still effective, resulting in no
comple-homolog of the human deafness geneMITF, affect neuroepithe-mentation.
lial and neural crest-derived melanocytes differently. Mech. Dev.
Our model predicts that gene expression is affected 70:155–166.
Pirrotta, V., 1999 Transvection and chromosomal
trans-interac-differently by the two versions of the MitfMi-wh protein
tion effects. Biochim. Biophys. Acta1424:M1–M8. than by the corresponding versions of wild-type Mitf
Raz, E., E. SchejterandB. Z. Shilo, 1991 Interallelic
complemen-proteins. Gene expression studies using gene arrays tation among DER/flballeles: implications for the mechanisms
of signal transduction by receptor-tyrosine kinases. Genetics129:
and/or studies on the effects of the different Mitf
pro-191–201. teins on well-defined promoter elements in the different
Schaible, R. H., 1963 Developmental genetics of spotting patterns
cell types can therefore be used to test the model in in the mouse. Ph.D. Thesis, Iowa State University, Ames, IA.
Steingrı´msson, E., K. J. Moore, M. L. Lamoreux, A. R. Ferre´
-vitro.
D’Amare´, S. K. Burleyet al., 1994 Molecular basis of mouse
We thank Debbie Swing, Joanne Dietz, and Fran Dorsey for expert microphthalmia(mi) mutations helps explain their developmental technical assistance and the staff of the Histopathology Laboratory, and phenotypic consequences. Nat. Genet.8:256–263. Frederick, Maryland, for help with histology. This work was supported Steingrı´msson, E., A. Nii, D. E. Fisher, A. R. Ferre´-D’Amare´, R. J.
McCormicket al., 1996 The semidominant Mi-b mutation
iden-by the National Cancer Institute (N.G.C. and N.A.J.), the National
tifies a role for the HLH domain in DNA binding in addition to Institute for Neurological Disorders and Stroke, DHHS (H.A.), and
its role in protein dimerization. EMBO J.15:6280–6289. the Icelandic Research Council (E.S. and J.H.H.).
Steingrı´msson, E., L. Tessarollo, B. Pathak, L. Hou, H.
Arn-heiteret al., 2002 Mitf and Tfe3, two members of the Mitf-Tfe
family of bHLH-Zip transcription factors, have important but functionally redundant roles in osteoclast development. Proc.
LITERATURE CITED
Natl. Acad. Sci. USA99:4477–4482.
Takebayashi, K., K. Chida, I. Tsukumoto, E. Morii, H. Munakata
Aroian, R. V., G. M.Lesaand P. W.Sternberg, 1994 Mutations in
et al., 1996 The recessive phenotype displayed by a dominant the Caenorhabditis eleganslet-23EGFR-like gene define elements
negativemirophthalmia-associated transcription factor mutant is important for cell-type specificity and function. EMBO J. 13:
360–366. a result of impaired nuclear localization potential. Mol. Cell. Biol.
Bentley, N. J., T. EisenandC. R. Goding, 1994 Melanocyte-specific 16:1203–1211.
expression of the human tyrosinase promoter: activation by the West, J. D., G. Fisher, J. F. Loutit, M. J. Marshall, N. W. Nisbet microphthalmia gene product and role of the initiator. Mol. Cell. et al., 1985 A new allele of microphthalmia induced in the
Biol.14:7996–8006. mouse: microphthalmia-defective iris (mi di). Genet. Res. 46:
Gru¨ neberg, H., 1953 The relations of microphthalmia and white 309–324.
in the mouse. J. Genet.51:359–362. Wolfe, H. G., andD. L. Coleman, 1964 Mi-spotted: a mutation in
Hallsson, J. H., J. Favor, C. Hodgkinson, T. Glaser, M. L. Lamor- the mouse. Genet. Res.5:432–440.
euxet al., 2000 Genomic, transcriptional and mutational analy- Yajima, I., S. Sato, T. Kimura, K. Yasumoto, S. Shibaharaet al., sis of the mouse microphthalmia locus. Genetics155:291–300. 1999 An L1 element intronic insertion in theblack-eyed white
Hemesath, T. J., E. Steingrı´msson, G. McGill, M. J. Hansen, J. (Mitfmi-bw) gene: the loss of a single Mitf isoform responsible for
Vaughtet al., 1994 microphthalmia, a critical factor in melano- the pigmentary defect and inner ear deafness. Hum. Mol. Genet.
cyte development, defines a discrete transcription factor family. 8:1431–1441.
Genes Dev.8:2770–2780. Yasumoto, K., andS. Shibahara, 1997 Molecular cloning of a cDNA
Hodgkinson, C. A., K. J. Moore, A. Nakayama, E. Steingrı´msson, encoding a human TFEC isoform, a newly identified
transcrip-N. G. Copelandet al., 1993 Mutations at the mouse microph- tional regulator. Biochim. Biophys. Acta1353:23–31.
thalmia locus are associated with defects in a gene encoding a Yasumoto, K., K. Yokoyama, K. Shibata, Y. TomitaandS. Shiba-novel basic-helix-loop-helix-zipper protein. Cell74:395–404. hara, 1994 Microphthalmia-associated transcription factor as
Hollander, W. F., 1964 Mouse News Lett.30:29. a regulator for melanocyte-specific transcription of the human
Hollander, W. F., 1968 Complementary alleles at the mi-locus in tyrosinase gene. Mol. Cell. Biol.14:8058–8070.
the mouse. Genetics60:189.