Dengue Virus Subverts Host Innate Immunity by Targeting Adaptor
Protein MAVS
Zhenjian He,a,b,cXun Zhu,b,c,dWeitao Wen,b,c,dJie Yuan,b,c,eYiwen Hu,b,c,dJiahui Chen,b,c,dShu An,b,c,dXinhuai Dong,b,c,d Cuiji Lin,b,c,dJianchen Yu,b,c,dJueheng Wu,b,c,dYi Yang,b,fJunchao Cai,b,c,dJun Li,b,c,eMengfeng Lib,c,d
School of Public Health, Sun Yat-sen University, Guangzhou, Chinaa
; Key Laboratory of Tropical Disease Control (Sun Yat-sen University), Ministry of Education, Guangzhou, Chinab
; Guangdong Province Key Laboratory of Functional Molecules in Oceanic Microorganism (Sun Yat-sen University), Bureau of Education, Guangzhou, Chinac
; Department of Microbiology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, Chinad
; Department of Biochemistry, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, Chinae
; Department of Pharmacology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, Chinaf
ABSTRACT
Dengue virus (DENV) is the most common mosquito-borne virus infecting humans and is currently a serious global health
chal-lenge. To establish infection in its host cells, DENV must subvert the production and/or antiviral effects of interferon (IFN). The
aim of this study was to understand the mechanisms by which DENV suppresses IFN production. We determined that DENV
NS4A interacts with mitochondrial antiviral signaling protein (MAVS), which was previously found to activate NF-
B and IFN
regulatory factor 3 (IRF3), thus inducing type I IFN in the mitochondrion-associated endoplasmic reticulum membranes
(MAMs). We further demonstrated that NS4A is associated with the N-terminal CARD-like (CL) domain and the C-terminal
transmembrane (TM) domain of MAVS. This association prevented the binding of MAVS to RIG-I, resulting in the repression of
RIG-I-induced IRF3 activation and, consequently, the abrogation of IFN production. Collectively, our findings illustrate a new
molecular mechanism by which DENV evades the host immune system and suggest new targets for anti-DENV strategies.
IMPORTANCE
Type I interferon (IFN) constitutes the first line of host defense against invading viruses. To successfully establish infection,
den-gue virus (DENV) must counteract either the production or the function of IFN. The mechanism by which DENV suppresses
IFN production is poorly understood and characterized. In this study, we demonstrate that the DENV NS4A protein plays an
important role in suppressing interferon production through binding MAVS and disrupting the RIG-I–MAVS interaction in
mitochondrion-associated endoplasmic reticulum membranes (MAMs). Our study reveals that MAVS is a novel host target of
NS4A and provides a molecular mechanism for DENV evasion of the host innate immune response. These findings have
impor-tant implications for understanding the pathogenesis of DENV and may provide new insights into using NS4A as a therapeutic
and/or prevention target.
D
engue virus (DENV) (family
Flaviviridae
, genus
Flavivirus
) is
an enveloped virus with a positive-sense, single-stranded
RNA genome that is responsible for dengue fever (DF) and the
more severe, life-threatening dengue hemorrhagic fever (DHF)
and dengue shock syndrome (DSS) (
1
). It has been estimated that
50 million to 100 million people are infected by DENV worldwide
each year, and 500,000 patients progress to severe DHF (
2
). DENV
is categorized into four antigenically related but distinct serotypes,
designated DENV1, -2, -3, and -4. The DENV genome is
⬃
11 kb
in length and encodes a single polyprotein that can be processed
co- and posttranslationally by cellular and viral proteases into
three structural (C, prM, and E) and seven nonstructural (NS1,
NS2A, NS2B, NS3, NS4A, NS4B, and NS5) proteins in the
endo-plasmic reticulum (ER) (
3
).
As an early response to viral infection, mammalian cells
pro-duce type I interferons (IFNs), mainly IFN-
␣
and IFN-

, which
repress virus replication (
4
,
5
). This early innate antiviral response
is initiated by viral recognition through pathogen recognition
re-ceptors (PRRs), including Toll-like rere-ceptors (TLRs) and retinoic
acid-inducible gene I (RIG-I)-like receptors (RLRs). RIG-I, a
member of the RLRs, binds viral RNA, which triggers
conforma-tional changes that expose CARD domains for subsequent
signal-ing (
6
). Signaling by RIG-I requires the adaptor protein
desig-nated mitochondrial antiviral signaling adaptor (MAVS) (also
known as IPS-1/VISA/Cardif) (
7–10
). MAVS then transduces
sig-nals from RIG-I through CARD-CARD domain interactions,
which activate IFN regulatory factor 3 (IRF3) and NF-
B through
a signaling cascade involving the cytosolic protein kinases
TANK-binding kinase 1 (TBK1) and I
B kinase
ε
(IKK
ε
) (
11
),
conse-quently leading to type I IFN production.
Previous studies have shown that DENV infection usually leads
to low levels of IFN-
␣
and -

, thus suggesting that DENV may
inhibit IFN production after DENV infection (
12–16
). Several
re-search groups have reported that prevention of STAT1
phosphor-ylation or induction of STAT2 degradation is a mechanism
possi-bly underlying the antagonistic effects of DENV on IFN signaling
Received5 February 2016Accepted23 May 2016
Accepted manuscript posted online1 June 2016
CitationHe Z, Zhu X, Wen W, Yuan J, Hu Y, Chen J, An S, Dong X, Lin C, Yu J, Wu J, Yang Y, Cai J, Li J, Li M. 2016. Dengue virus subverts host innate immunity by targeting adaptor protein MAVS. J Virol 90:7219 –7230.doi:10.1128/JVI.00221-16.
Editor:J. U. Jung, University of Southern California
Address correspondence to Mengfeng Li, [email protected]. Z.H., X.Z., and W.W. contributed equally to this work.
Supplemental material for this article may be found athttp://dx.doi.org/10.1128 /JVI.00221-16.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
crossmark
on November 7, 2019 by guest
http://jvi.asm.org/
(
17–22
). However, previous studies focused mainly on the effects
of DENV on antiviral signaling downstream of IFN, leaving the
molecular mechanism underlying the suppression of IFN
produc-tion by DENV largely unknown. Recently, two groups reported
similar results showing that human mediator of IRF3 activation
(MITA) or stimulator of interferon genes (STING) is cleaved by
the DENV protease NS2B/3 (
23
,
24
).
In the present study, we report that DENV NS4A potently
dis-rupts the induction of IFN by targeting MAVS. We found that
NS4A binds MAVS and prevents the binding of MAVS with
RIG-I, hence abrogating IFN induction. Furthermore, we
demon-strate that NS4A is associated with the N-terminal CARD-like
(CL) domain and the C-terminal transmembrane (TM) domain
of MAVS, and the third transmembrane (TM3) domain of NS4A
is essential for binding to MAVS. Thus, our findings collectively
suggest that NS4A contributes to DENV immune evasion through
the inhibition of MAVS-mediated cellular responses.
MATERIALS AND METHODS
Plasmids.A plasmid encoding human MAVS with an N-terminal Flag epitope tag was amplified by reverse transcription-PCR (RT-PCR) and cloned into the KpnI and XbaI sites of the pcDNA3.1 vector. The plasmid expressing Flag-tagged RIG-I was obtained from Addgene (Addgene plas-mid 27236). For the green fluorescent protein (GFP)-IRF3 construct, human IRF3 was amplified from plasmid pcDNA3-V5-IRF3 (Addgene plasmid 32713) and subcloned into the XhoI and EcoRI sites of the pEGFP-C3 vector. To generate plasmids pFN10A(ACT)-RIG-I, MAVS, TBK1, and pFN10A(ACT)-IKKε, each of the RIG-I, MAVS, TBK1, and IKKεfragments was amplified by PCR and subcloned into the pFN10A(ACT) vector (Promega, San Luis Obispo, CA), respectively. cDNA encoding each DENV protein, including C, prM, E, NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5, was amplified by RT-PCR using DENV2 strain New Guinea C (NGC) cDNA as a tem-plate and cloned into the pFN11A(BIND) vector (Promega, San Luis Obispo, CA). Truncated forms of MAVS (amino acids [aa] 1 to 77, 74 to 173, and 174 to 540) with N-terminal Flag epitope tags were amplified from the full-length template and cloned into the pcDNA3.1 vector. Plas-mids expressing Myc-tagged full-length NS4A and deletion mutants (aa 1 to 49, 1 to 74, 1 to 100, and 1 to 127) were also amplified by PCR and cloned into the pcDNA3.1 vector. Plasmids encoding DENV2 prM and influenza virus PB1-F2 (A/Puerto Rico/8/1934 [PR8] strain) with N-ter-minal Myc epitope tags were amplified by RT-PCR and cloned into the KpnI and BamHI sites of the pcDNA3.1 vector. Plasmid pEF-BOS-NS4A with a C-terminal 6⫻His tag was constructed by cloning the NS4A frag-ment into the pEF-BOS vector using the XhoI and BamHI sites. The pIFN--luc reporter plasmid was constructed by cloning a 125-bp frag-ment of the IFN-promoter into the pGL3-Basic vector using the NheI and HindIII sites, as previously described (25). All constructs were veri-fied by DNA sequencing. The PCR primers used in this study are summa-rized in Table S1 in the supplemental material.
Cell culture and virus.HeLa (purchased from ATCC) and 293T (ob-tained from the Cell Bank of the Chinese Academy of Sciences, Shanghai, China) cells were cultured at 37°C with 5% CO2in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Carlsbad, CA), 2 mML-glutamine, 100g/ml streptomycin, and 100 U/ml penicillin (Invitrogen, Carlsbad, CA).Aedes albopictusC6/36 cells (ATCC CRL-1660) (26) were main-tained at 28°C with 5% CO2in DMEM supplemented with 10% FBS.
DENV2 strain NGC (GenBank accession numberM29095) was kindly provided by the Guangzhou Center for Disease Control and Prevention (CDC) (27) and propagated in the mosquito cell line C6/36. Virus stocks were titrated by fluorescence-activated cell sorter (FACS) assays with C6/36 cells according to a previously described method (28). Sendai virus
(SeV) was grown in 10-day-old embryonated chicken eggs and titrated by a hemagglutination assay as previously described (29,30).
Luciferase reporter assays.293T cells seeded into 24-well plates were transiently transfected with plasmids encoding IFN-and the internal control pRL-TK together with NS4A (250 and 500 ng), prM (500 ng), or PB1-F2 (500 ng). Cells were then infected with SeV at 100 hemagglutinat-ing units (HAU)/ml for 16 h, followed by analysis of cell lysates for lucif-erase activity with a Dual-Luciflucif-erase Reporter Assay System kit (Promega, San Luis Obispo, CA) according to the manufacturer’s protocol.
Mammalian two-hybrid assay.293T cells were seeded into a 24-well plate 24 h prior to transfection. Next, 100 ng (each) of the pFN11A(BIND) vector expressing an individual DENV protein with a GAL4 DNA binding domain (GAL4-BD) fusion protein and 100 ng (each) of the pFN10A(ACT) vector expressing the RIG-I, MAVS, TBK1, or IKKε pro-tein was cotransfected with 250 ng of reporter plasmid pGL4.31 into 293T cells by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The pFN11A(BIND) vector contained aRenillaluciferase gene, which was used as an internal control to normalize the DNA transfection efficiency. The pBIND and pACT vectors were used as the negative controls, and the pBIND-Id and pACT-MyoD vectors were used as the positive controls, according to the manufacturer’s instructions (Promega, San Luis Obispo, CA). After 48 h, firefly andRenillaluciferase activities were determined by using a Dual-Luciferase Reporter Assay System kit (Promega, San Luis Obispo, CA).
Western blotting.Cells were lysed with sampling buffer (50 mM Tris-HCl [pH 7.4], 1 mM phenylmethylsulfonyl fluoride [PMSF], 10% glyc-erol, 6% SDS, 5% beta-mercaptoethanol, and 0.1% bromophenol blue), and protein concentrations were measured with a bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, Rockford, IL). Protein samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride (PVDF) membrane. Nonspecific antibody binding sites were blocked with 5% nonfat milk in Tris-buffered saline (TBS) (20 mM Tris-HCl [pH 7.6], 135 mM NaCl, and 0.1% Tween 20) for 1 h at room temperature and then reacted with the following primary antibodies: anti-MAVS (Bethyl Laboratories, Montgomery, TX), anti-anti-MAVS (T-20) (Santa Cruz Biotechnology, Santa Cruz, CA), anti-phospho-IRF3 (S396) (Cell Signaling, Danvers, MA), anti-IRF3 (Cell Signaling, Danvers, MA), anti-NS4A (GeneTex Inc., Irvine, CA), anti-Flag M2, anti-c-Myc, and anti--actin (Sigma-Aldrich, St. Louis, MO). Membranes were incubated with horseradish peroxidase-conjugated secondary antibody, and signals were detected by enhanced chemiluminescence using a commercial kit (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer’s suggested protocols.
Immunofluorescence assay.Cells were plated onto coverslips in a 24-well plate and transfected with the indicated plasmids (500 ng). At 24 h posttransfection, cells were washed once with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde in PBS. Cells were permeabil-ized with 0.2% Triton X-100 and blocked for 30 min at room temperature with 10% bovine serum albumin (BSA) in PBS, followed by incubation with the primary antibody for 1 h. After three washes with PBS containing 0.1% Tween 20 (PBST), cells were incubated with fluorescein isothiocya-nate (FITC)- or rhodamine-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) or with Alexa Flour 488 dye- and Alexa Flour 647 dye-conjugated secondary antibodies (Life Technologies, Grand Island, NY) for 30 min and then incubated with 4=,6-diamidino-2-phenylindole (DAPI) for 10 min. Finally, coverslips were washed extensively and fixed onto slides. Images were taken under an LSM780 confocal microscope (Carl Zeiss MicroImaging GmbH, Jena, Germany).
Coimmunoprecipitation assay.293T cells were transfected with the indicated plasmids (10g/ml) by using a standard calcium phosphate coprecipitation method. Twenty-four hours later, cells were lysed with protein lysis buffer containing 25 mM HEPES, 150 mM NaCl, 1 mM EDTA, 2% glycerol, 1% NP-40, and a cocktail of protease and
phospha-He et al.
on November 7, 2019 by guest
http://jvi.asm.org/
tase inhibitors (Roche, Basel, Switzerland). Lysates were incubated with the indicated antibodies or IgG antibody overnight at 4°C. Subsequently, precipitates were washed five times with wash buffer containing 20 mM HEPES, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2% glycerol, and 0.1% NP-40; resuspended in sampling buffer; and examined by Western blot-ting. For the endogenous immunoprecipitation (IP) experiment, lysates were incubated with the indicated antibodies, and protein G Dynabeads (Life Technologies, Grand Island, NY) were subsequently added to cap-ture the protein complexes according to the manufaccap-turer’s suggested protocol.
Biacore analysis.Surface plasmon resonance (SPR) data were ob-tained with a Biacore T100 instrument and research-grade CM5 sensor chips (General Electric Company [GE]) according to the manufacturer’s protocol. Briefly, MAVS was immobilized on a CM5 sensor chip. Differ-ent concDiffer-entrations (0, 62.5, 125, 250, 500, 1,000, and 2,000 nM) of NS4A were injected at a flow rate of 30l/min for 3 min. Association data were collected for 3 min, followed by a 20-min dissociation period. The binding surfaces were regenerated after each injection by injection of 10l of 10 mM NaOH for 20 s. All procedures were performed at 25°C by using standard HBS (HEPES-buffered saline; GE) as a running buffer. A buffer injection control was included for each experiment and was subtracted from the data before analysis. The equilibrium dissociation constant (KD)
was calculated as previously described (31). Data analysis to obtain bind-ing curves was performed by usbind-ing BIAevaluation 3.1 (Biacore), by glob-ally fitting the data to a simple 1:1 Langmuir ligand binding model defined by the formula Bound⫽ (Bmax⫻ [NS4A])/(KD ⫻[NS4A]), where
“Bound” is measured in response units (RU),Bmaxis the maximum
re-sponse (in rere-sponse units), and [NS4A] is the concentration of the NS4A protein. Recombinant human MAVS protein was purchased from OriGene (catalog no. TP308175). Recombinant NS4A was produced from 293T cells and purified by using Ni-nitrilotriacetic acid (NTA) affinity chromatography (QIAexpressionist; Qiagen, Chatsworth, CA), followed by Superdex 200 (GE) gel filtration chromatography, as previously de-scribed (32,33).
Real-time PCR.Total cellular RNA was prepared with TRIzol (Invit-rogen, Carlsbad, CA) according to the manufacturer’s instructions. First-strand cDNA synthesis was performed by using random hexamer primers, and real-time PCR was carried out by using FastStart Universal SYBR green master mix (Roche, Basel, Switzerland). All readings were normal-ized to the level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. The primer sets used for real-time PCR are shown in Table S1 in the supplemental material.
Isolation of monocytes.Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by Ficoll-Hypaque density gradient cen-trifugation according to the manufacturer’s instructions (Lymphoprep; Nycomed, Oslo, Norway). Primary monocytes were purified from PBMCs by using Monocyte Isolation kit II (Miltenyi Biotec GmbH, Ber-gisch Gladbach, Germany) according to the protocol provided by the manufacturer. Purified monocytes were resuspended and cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% FBS (HyClone, Logan, UT), 15 mM HEPES, 2 mML-glutamine, 100
g/ml streptomycin, and 100 U/ml penicillin (Invitrogen, Carlsbad, CA).
Subcellular fractionation. Mitochondrion-associated endoplasmic reticulum membranes (MAMs), mitochondria, and cytosolic fractions containing lysosomes and microsomes were isolated from primary mono-cytes or 293T cells by using Percoll gradient fractionation as described previously (34). Equivalent amounts of protein from each fraction were determined by Western blotting.
Statistical analysis.The results are expressed as means⫾standard deviations (SD). Statistical analyses were performed on data from tripli-cate experiments by using two-tailed Student’sttest.
RESULTS
DENV NS4A interacts with MAVS, which mediates the type I
IFN induction pathway.
To investigate whether DENV proteins
interfere with the IFN induction pathway by interacting with the
pathway components, we employed the mammalian two-hybrid
system to identify molecular targets of DENV proteins in PRR
signaling. Each of the 10 DENV proteins was expressed as a fusion
protein with the yeast GAL4-BD, and the 4 key PRR signaling
molecules, i.e., RIG-I, MAVS, TBK1, and IKK
ε
, were fused to the
transcriptional activation domain of herpes simplex virus VP16
(VP16-AD). 293T cells were then cotransfected with 10 DENV
proteins and the 4 key PRR signaling proteins, and the expression
of each transfected gene was verified by Western blotting using
available commercial antibodies or antibodies against the Flag,
Myc, or hemagglutinin (HA) tag (see Fig. S1 in the supplemental
material). As shown in
Fig. 1
, there was no interaction between
DENV proteins and RIG-I, TBK1, or IKK
ε
(
Fig. 1A
,
C
, and
D
),
whereas MAVS and NS4A interacted with each other (
Fig. 1B
),
suggesting that MAVS may be an interactive partner of DENV
NS4A. Furthermore, we employed an IFN-

luciferase reporter
assay to determine whether NS4A could reverse the activation of
the IFN-

promoter driven by MAVS. Overexpression of the
NS4A and MAVS proteins was confirmed by Western blotting
(
Fig. 1E
). Our results showed that DENV2 NS4A markedly
de-creased the reporter luciferase activity induced by MAVS (
Fig.
1E
). To further test whether the NS4A-MAVS interaction might
also function to inhibit type I IFN production, we examined the
effects of the DENV3 and DENV4 NS4A proteins on IFN-

re-porter activity, and our results showed that the NS4A proteins of
both DENV3 and DENV4 had inhibitory effects on IFN-

re-porter activities driven by MAVS (
Fig. 1F
), indicating that the
identified NS4A-MAVS interaction represents a common
anti-IFN mechanism across different serotypes of dengue virus.
DENV NS4A physically colocalizes with MAVS during
DENV infection.
Based on the finding that NS4A interacts with
MAVS, we next asked whether the two molecules colocalize. For this
purpose, HeLa cells were infected with DENV2 and then stained for
NS4A and MAVS. As shown in
Fig. 2A
, confocal microscopy revealed
costaining of NS4A and MAVS in cells infected by DENV2,
suggest-ing colocalization of the two proteins (
Fig. 2A
). We next used the
DENV protein prM, which did not interact with MAVS in the
mam-malian two-hybrid assay (
Fig. 1B
), as a negative control and the
in-fluenza virus PB1-F2 protein, which has been reported to antagonize
type I IFN induction by targeting MAVS, as a positive control (
35
,
36
). Our results showed that PB1-F2, but not DENV prM, also
colo-calized with MAVS (
Fig. 2A
).
To address whether the NS4A protein physically interacts with
MAVS, we performed endogenous IP assays on DENV2-infected
cell lysates. Our results showed that NS4A, but not prM, could be
precipitated by the MAVS antibody (
Fig. 2B
), suggesting that
NS4A and MAVS form a complex in DENV-infected cells. To
better elucidate the interaction between NS4A and MAVS, we
per-formed
in vitro
co-IP assays based on MAVS and NS4A
overex-pression, and the results showed that MAVS interacted with NS4A
(
Fig. 2C
). Additionally, we performed IP to precipitate
endoge-nous MAVS using Myc-tagged NS4A and cells transfected with
prM or PB1-F2 as controls. We found that endogenous MAVS was
pulled down together with Myc-tagged NS4A or PB1-F2 but not
prM (
Fig. 2D
and
E
), further confirming the ability of DENV
NS4A to form a complex with MAVS. The binding affinities
be-tween MAVS and NS4A were also analyzed by Biacore surface
plasmon resonance assays with MAVS immobilized on the chip
surface and injections containing analytes consisting of serial
di-Targeting MAVS by Dengue Virus NS4A
on November 7, 2019 by guest
http://jvi.asm.org/
lutions of NS4A proteins. The data showed a significant increase
in NS4A binding to MAVS in solution, with an apparent
KD
of
1.12
⫻
10
⫺8M (
Fig. 2F
). These data indicate that NS4A directly
interacts with MAVS.
DENV NS4A translocates to the MAM and interacts with
MAVS.
The ER contains specialized intracellular components,
known as MAMs, that physically connect the ER to mitochondria
(
37
,
38
). Because DENV NS4A is an ER membrane-bound protein
FIG 1Mammalian two-hybrid analyses of interactions between DENV proteins and IFN signaling proteins. Protein-protein interactions were determined by the mammalian two-hybrid system. (A to D) 293T cells in 24-well plates were cotransfected with a pGL4.31 vector, a pFN11A(BIND) vector expressing a fusion protein of the GAL4-BD and individual DENV proteins, and a pFN10A(ACT) vector expressing the RIG-I (A), MAVS (B), TBK1 (C), or IKKε(D) protein. The pFN11A(BIND) vector contained aRenillaluciferase gene that was used as an internal control to normalize DNA transfection efficiency. At 48 h posttransfection, the levels of luciferase activity were examined. The results are shown as relative luciferase activity after normalization toRenillaluciferase activity. The pBIND and pACT vectors were used as negative controls, and the pBIND-Id and pACT-MyoD vectors were used as positive controls (PC), according to the manufacturer’s instructions. (E and F) Luciferase activities of 293T cells transfected with an IFN-luciferase reporter, the pRL-TK (internal control) reporter, and the plasmid expressing MAVS. An empty vector or a vector expressing DENV2 NS4A (500 ng) (E), DENV3 NS4A (500 ng) (F), or DENV4 NS4A (500 ng) (F) was also cotransfected. At 36 h posttransfection, cell lysates were harvested for luciferase activity assays. Overexpression of NS4A and MAVS proteins was confirmed by Western blotting. The data are shown as the means⫾SD derived from three repeat experiments. ** indicates aPvalue of⬍0.01 (Student’sttest). RLU, relative luciferase units.
He et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.81.502.65.570.2]FIG 2DENV NS4A colocalizes and interacts with MAVS during DENV infection. (A) HeLa cells infected with DENV2 at an MOI of 10 or mock-infected HeLa cells were fixed with 4% paraformaldehyde and costained with anti-MAVS and anti-NS4A or anti-prM antibodies as well as DAPI. HeLa cells transfected with a plasmid expressing Myc-tagged influenza virus PB1-F2 (500 ng) or an empty vector were stained with anti-Myc and anti-MAVS antibodies as well as DAPI. HeLa cells expressing Myc-tagged NS4A (500 ng) were stained with anti-Myc antibody and an antibody against the ER marker calnexin. Secondary antibodies conjugated to rhodamine and Alexa Fluor 488 dye were used to visualize the indicated proteins. Images are representative of results from three independent experiments, and a semiquantitative colocalization analysis of the indicated proteins was performed by using the line profile of ZEN imaging software. Fluorescence intensities along the red lines sectioning the cells are plotted for the indicated proteins. (B) 293T cells were infected with DENV2 at an MOI of 10, followed by immunoprecipitation using anti-MAVS antibody or control mouse IgG, and protein G Dynabeads were added to capture the immunocomplexes. The immunocomplexes were analyzed by Western blotting using anti-NS4A, anti-prM, or anti-MAVS antibodies (Abs). The arrow indicates the position of NS4A, and IgG-L indicates the IgG light chain. IB, immunoblotting. (C) 293T cells cotransfected with plasmids encoding Flag-tagged MAVS (10g/ml) and Myc-tagged NS4A (10g/ml) were used in a co-IP assay. Cell lysates were precipitated with anti-Flag antibody or control mouse IgG, and immunocomplexes were analyzed with the indicated antibodies by Western blotting. (D and E) 293T cells transfected with plasmids encoding Myc-tagged NS4A (D) or prM or PB1-F2 (E) (10g/ml) were used for precipitation experiments with anti-Myc antibody or control mouse IgG, and immunocomplexes were determined with anti-MAVS antibody by Western blotting. A small aliquot of the cell lysate was also prepared before IP for equal input of Flag-MAVS, Myc-NS4A, Myc-prM, or Myc–PB1-F2. (F) SPR analysis of interactions between MAVS and NS4A. Direct binding was measured by Biacore assays. MAVS was immobilized on a CM5 chip. The analytes consisted of serial dilutions of NS4A proteins at concentrations of between 0 nM and 2,000 nM. The data shown are representative of data from three independent experiments with similar results.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.38.540.48.561.2](
39
), we assessed whether MAMs were able to position DENV
NS4A in proximity to MAVS and thus facilitate their interaction.
To address this issue, HeLa cells overexpressing Myc-tagged NS4A
were examined for the localization pattern of MAVS and NS4A.
Calnexin and MitoTracker Red were used as ER and
mitochon-drion markers, respectively, and MAMs and the peroxisome were
labeled by antibodies against fatty acid-coenzyme A ligase 4
(FACL4) and the 70-kDa peroxisomal membrane protein
(PMP70), respectively. We found that a significant fraction of
NS4A colocalized with MAVS in the MAM, and a small fraction of
NS4A colocalized with MAVS in the mitochondria but not in the
peroxisome (
Fig. 3A
to
C
). Moreover, when we isolated MAMs
from primary monocytes infected with DENV2 and performed
Western blotting to analyze NS4A, we detected both NS4A and the
MAM marker FACL4 in the MAM fraction (
Fig. 3D
). We also
isolated MAMs from 293T cells transfected with Myc-tagged
NS4A and detected Myc-tagged NS4A in the MAM fractions (
Fig.
3E
). Together, these data suggested that NS4A may translocate to
the MAM and interact with MAVS via this platform.
DENV infection and NS4A prevent RIG-I from forming
complexes with MAVS.
MAVS transduces signals from RIG-I
through CARD-CARD domain interactions, leading to the
ac-FIG 3DENV NS4A translocates to the MAM and interacts with MAVS. (A) Following transfection with Myc-tagged NS4A (500 ng) or an empty vector, HeLa cells were fixed with 4% paraformaldehyde and stained with anti-Myc antibody for NS4A, anti-FACL4 antibody for MAMs, and an anti-MAVS antibody. Secondary antibodies conjugated to Alexa Fluor 488, rhodamine, and Alexa Fluor 647 were used to visualize the stained NS4A, FACL4, and MAVS proteins, respectively. All images for all panels are representative of results from three independent experiments, and a semiquantitative colocal-ization analysis of the indicated proteins was performed by using the line profile of ZEN imaging software. Fluorescence intensities along the red lines sectioning the cells are plotted for the indicated proteins. (B) HeLa cells were transfected with a Myc-tagged NS4A expression plasmid (500 ng) or an empty vector. At 24 h posttransfection, cells were incubated with MitoTracker Red dye, fixed with 4% paraformaldehyde, and then stained with anti-Myc and anti-MAVS antibodies as well as DAPI, followed by secondary antibodies conjugated to Alexa Fluor 488 and Alexa Fluor 647 to visualize the NS4A and MAVS proteins, respectively. (C) HeLa cells expressing Myc-tagged NS4A (500 ng) or an empty vector were stained with anti-Myc for NS4A, anti-PMP70 antibody for the peroxisome, and anti-MAVS antibody. Secondary antibodies conjugated to rhodamine, Alexa Fluor 488, and Alexa Fluor 647 were used to visualize stained NS4A, PMP70, and MAVS proteins, respectively. (D) Primary monocytes that were infected with DENV2 at an MOI of 10 or mock infected were used to isolate a MAM, and the isolated MAM was analyzed by Western blotting. (E) 293T cells transfected with Myc-tagged NS4A (10g/ml) were used to isolate MAMs and were analyzed by Western blotting. WCL, whole-cell lysates; Cyto, cytosol. The Western blot shown is representative of data from three independent experiments with similar results.
He et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.135.455.69.443.2]tivation of IRF3 and IFN induction (
11
). To investigate
whether the suppressed binding of endogenous RIG-I to MAVS
contributes to the inhibition of IFN by DENV infection, we
examined the formation of RIG-I–MAVS complexes after
DENV infection. Primary monocytes were infected with
DENV2 at different multiplicities of infection (MOIs) or mock
infected for 24 h and then stimulated with SeV for 16 h. Cell
lysates were harvested, immunoprecipitated with an
anti-MAVS antibody, and analyzed by Western blotting using an
anti-RIG-I antibody. As shown in
Fig. 4A
, RIG-I interacted
with MAVS following SeV stimulation, but cells preinfected
with DENV2 exhibited a significant decrease in the interaction
between RIG-I and MAVS, and such an inhibition correlated
with the MOI of DENV2 used for infection. To address whether
NS4A affects the interaction between RIG-I and MAVS, we
examined the formation of RIG-I–MAVS complexes in the
presence or absence of NS4A. 293T cells were transfected with
different amounts of Myc-tagged NS4A or the control vector
and then stimulated with SeV prior to IP assays. Our results
showed that cells expressing NS4A exhibited a significantly
re-pressed interaction between RIG-I and MAVS in a
dose-depen-dent manner (
Fig. 4B
). Together, these data indicate that NS4A
binds MAVS, which may in turn prevent its interaction with
RIG-I, modulate downstream signaling, and suppress IFN
pro-duction.
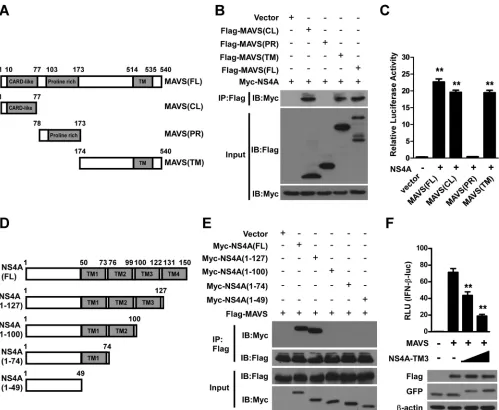
NS4A interacts with both the CL and TM domains of MAVS.
MAVS is a 540-aa protein consisting of three structural and
functional domains, namely, the N-terminal CARD-like (CL)
domain, the middle proline-rich (PR) domain, and the
C-ter-minal transmembrane (TM) domain (
Fig. 5A
). To map the
MAVS domains involved in NS4A binding, we generated the
following serial MAVS deletion mutants: Flag-MAVS(CL),
ex-pressing aa 1 to 77 and containing the CARD-like domain;
Flag-MAVS(PR), expressing aa 78 to 173 and containing the
proline-rich domain; Flag-MAVS(TM), expressing aa 174 to
540 and containing the transmembrane domain; and
Flag-MA-VS(FL), expressing full-length MAVS (
Fig. 5A
). Co-IP assays
showed that either the CL or TM domain of MAVS could pull down
NS4A (
Fig. 5B
). Furthermore, the results of mammalian two-hybrid
experiments also confirmed that the CL or TM domain of MAVS
could interact with NS4A (
Fig. 5C
). Taken together, these results
sug-gest that both domains contribute to the formation of the
MAVS-NS4A complex.
The TM3 domain of NS4A is required for interaction with
MAVS.
To identify the NS4A region(s) required for binding with
MAVS, we performed serial deletion mutagenesis analysis of
NS4A (
Fig. 5D
). Myc-tagged full-length NS4A and four deletion
constructs of NS4A were cotransfected with Flag-tagged MAVS.
The subsequent co-IP assays showed that NS4A lacking the
C-ter-minal TM4 domain was still able to interact with MAVS, similarly
to full-length NS4A (
Fig. 5E
). However, further deletion of the
TM3 domain completely abrogated the interaction between NS4A
and MAVS (
Fig. 5E
), suggesting that the TM3 domain is critical
for NS4A to bind MAVS. Next, we asked whether the TM3
do-main has an inhibitory effect on MAVS, and an IFN-

luciferase
reporter assay was performed. Our results showed that the TM3
domain alone markedly decreased reporter luciferase activity
driven by MAVS (
Fig. 5F
), further demonstrating that the
C-ter-minal TM3 domain of NS4A is required for a specific
NS4A-MAVS interaction.
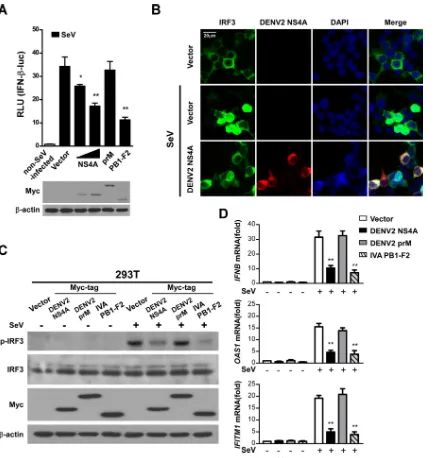
DENV NS4A downregulates type I IFN production by
inhib-iting IRF3 activation.
To further investigate how NS4A inhibits
the signaling pathway mediating type I IFN production, we used
SeV as a stimulus and determined the effect of NS4A on IFN-

luciferase reporter activity. Our results showed that both NS4A
and PB1-F2, but not prM, inhibited SeV-induced activation of the
reporter. Overexpression of the NS4A, prM, or PB1-F2 protein
was confirmed by Western blotting (
Fig. 6A
).
The transcription factor IRF3 is a key innate immune system
component that mediates IFN-

induction. Once IRF3 is
phos-phorylated, it forms a dimer, translocates into the nucleus from
the cytoplasm, and induces the expression of IFN-

and
IFN-stimulated genes (ISGs) through specifically binding to their
pro-moter regions (
40
). To further investigate how NS4A inhibits the
signaling that mediates type I IFN production, we assessed the
translocation of IRF3 in 293T cells that expressed GFP-tagged
IRF3 or NS4A following infection with SeV. In cells transfected
with a blank control vector, 32.46% (
⫾
3.85%) of IRF3 rapidly
FIG 4DENV2 infection or ectopic NS4A prevents RIG-I from binding MAVS. (A) Primary monocytes were infected with DENV2 at the indicated MOIs (0, 5, and 10) or mock infected for 24 h and then infected with SeV for 16 h. Cell lysates were harvested, subjected to immunoprecipitation using an anti-MAVS antibody, and analyzed by Western blotting. (B) 293T cells were transfected with different amounts of Myc-tagged NS4A (0, 10, and 20g/ml) or a blank vector control for 24 h and then infected with SeV for 16 h. Whole-cell lysates were subjected to immunoprecipitation using an anti-MAVS antibody and analyzed by Western blotting. Expression of precipitated proteins was determined by Western blotting using the indicated antibodies (bottom). The data shown are representative of data from three independent experiments with similar results.
Targeting MAVS by Dengue Virus NS4A
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.97.496.64.212.2]translocated into the nucleus after SeV infection. In contrast, IRF3
was retained in the cytoplasm of cells expressing ectopic NS4A
after SeV stimulation, and only 4.91% (
⫾
0.66%) of IRF3 was
translocated into the nucleus (
Fig. 6B
), indicating a suppressive
function of NS4A in IRF3 translocation. We next determined the
phosphorylation of endogenous IRF3 in cells with or without the
expression of NS4A. Our data showed that SeV-induced
phos-phorylation of IRF3 was repressed by NS4A or PB1-F2 but not
prM (
Fig. 6C
). Additionally, NS4A or PB1-F2, but not prM,
mark-edly diminished mRNA production of
IFNB
,
OAS1
, and
IFITM1
following SeV infection (
Fig. 6D
). These data demonstrate that
NS4A inhibits the expression of type I IFN induced by viral
infec-tion or MAVS through blocking the phosphorylainfec-tion and
trans-location of IRF3.
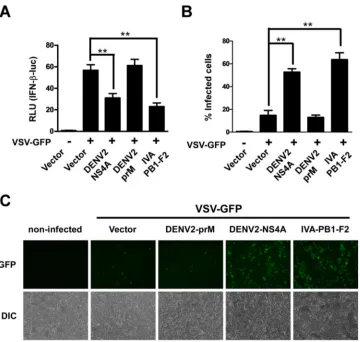
Previous studies have shown that a recombinant Newcastle
disease virus (NDV)-GFP or vesicular stomatitis virus (VSV)-GFP
FIG 5Both the CL and TM domains contribute to MAVS complex formation with NS4A, and the TM3 domain of NS4A is required for binding to MAVS. (A) Schematic diagram of the MAVS protein and the following functional domains: the CARD-like domain (aa 10 to 77), the proline-rich domain (aa 103 to 173), and the transmembrane domain (aa 514 to 535). (B) Co-IP and Western blotting of 293T cells transfected with Myc-tagged NS4A (10g/ml) along with vectors expressing the indicated Flag-tagged MAVS truncation forms or full-length MAVS (10g/ml). An empty vector was used as a negative control. (C) 293T cells in 24-well plates were cotransfected with the pGL4.31 vector; a pFN11A(BIND) vector expressing NS4A; and a pFN10A(ACT) vector expressing MAVS(FL), MAVS(CL), MAVS(PR), or MAVS(TM). At 48 h posttransfection, levels of luciferase activity were examined. pBIND and pACT vectors were used as negative controls. (D) Schematic illustration of the DENV NS4A protein and its serial deletion mutants. (E) 293T cells were cotransfected with a Flag-MAVS plasmid and individual Myc-tagged NS4A truncated plasmids or full-length NS4A (10g/ml). A blank vector served as a negative control. At 24 h posttransfection, cells were lysed, co-IP was performed by using the anti-Flag antibody, and immunocomplexes were analyzed by Western blotting. The presence of transfected proteins in cell lysates (input) was verified by detection with the indicated antibodies (bottom). The numbers at the top indicate amino acid positions. The data shown are representative of results from three independent experiments. (F) Luciferase activities of 293T cells were measured after transfection with an IFN-luciferase reporter and the pRL-TK reporter together with a plasmid expressing MAVS. A blank vector control or increasing amounts of a vector expressing the TM3 domain of NS4A (NS4A-TM3) (250 and 500 ng) were also cotransfected. At 36 h posttransfection, cell lysates were harvested for luciferase activity assays and Western blotting. Data are presented as the means⫾ SD derived from three repeat experiments. **,P⬍0.01 (Student’sttest).
He et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.44.543.64.474.2]system can be used as a strategy to screen proteins possessing
IFN-antagonizing activity (
41
,
42
). In the present study, we
em-ployed recombinant VSV-GFP to investigate whether DENV
NS4A is an antagonist of IFN production. When 293T cells
ex-pressed NS4A, a high level of VSV-GFP replication was present,
which was consistent with the notion that NS4A inhibits IFN
pro-duction in host cells (
Fig. 7A
to
C
). Upon VSV-GFP stimulation,
NS4A reduced IFN-

reporter activity, suggesting that the
inhib-itory effect of DENV NS4A on IFN production is also present
during actual viral infection.
DISCUSSION
The innate immune system is an evolutionarily conserved system
functioning as the first line of defense against invading pathogens
and is essential for the subsequent activation of adaptive
immu-nity. Central to early antiviral defense is the induction of IFN,
FIG 6NS4A negatively regulates the IFN signaling pathway by inhibiting IRF3 activation. (A) 293T cells transfected with the IFN-reporter together with NS4A (250 and 500 ng), prM (500 ng), or PB1-F2 (500 ng) were infected with SeV at 100 HAU/ml for 16 h, followed by analysis of cell lysates for luciferase activity. The protein expression levels of NS4A, prM, and PB1-F2 used in this reporter assay were determined by Western blotting. (B) Representative fluorescence micrographs of IRF3 in 293T cells transfected with GFP-IRF3 together with an empty vector or Myc-tagged NS4A (500 ng) for 24 h and then infected with SeV for 16 h. DAPI is a DNA-intercalating dye. (C) Western blotting of phosphorylated and total IRF3 expression in 293T cells transfected with a blank control vector or a vector expressing Myc-tagged NS4A, prM, or PB1-F2 and infected with SeV. (D)IFNB,OAS1, andIFITM1mRNA levels in 293T cells transfected with a blank control vector or a vector expressing Myc-tagged NS4A, prM, or PB1-F2 (1g/well) and then infected with SeV were determined by real-time PCR. IVA, influenza virus A. The data are presented as the means⫾SD derived from three repeat experiments. **,P⬍0.01 (Student’sttest).
Targeting MAVS by Dengue Virus NS4A
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.77.513.63.521.2]especially type I IFN, which further leads to the expression of a set
of antiviral ISGs (
43
). DENV is one of the most important
mos-quito-borne viruses affecting humans, causing dengue fever or
more severe dengue symptoms such as DHF and DSS. Like other
viruses, DENV evades host immune defenses. In the present study,
a novel function of NS4A—suppression of IFN-

by interacting
with MAVS and subsequently blocking IRF3 phosphorylation and
nuclear translocation— has been identified, highlighting an
im-portant role for NS4A in the establishment of DENV infection in
humans.
DENV is known to be a weak inducer of type I IFN, and
DENV-infected human dendritic cells exhibit an impaired type I IFN
response to infection with several viruses and to stimulation with
poly(I·C) (
14
,
44
,
45
). Thus far, the functional significance of
NS4A in viral replication remains poorly characterized. NS4A was
previously suggested to harbor viral replication complexes by
in-ducing curvature of the ER membrane and promoting membrane
rearrangements (
39
). Our findings reveal that NS4A may have
evolved to be a multifunctional protein that is directly involved in
the DENV replication machinery and also contributes to the
eva-sion of host immunity. It was demonstrated previously that
DENV NS2B/3 blocks the type I IFN response by targeting MITA/
STING (
23
,
24
). Our work extends these findings by showing that
NS4A proteins derived from various serotypes of DENV also
block type I IFN production through targeting MAVS-mediated
signaling pathways, implying that the inhibition of
MAVS-depen-dent IFN activation by DENV NS4A is not limited to only one
DENV serotype. Further investigation of how this effect of NS4A
cooperates with other counter-IFN effects of DENV, such as IFN
inhibition by NS2B/3 via proteolytic cleavage of MITA (
23
), is
warranted. The knowledge gained by investigating the
mecha-nisms of DENV immune evasion may facilitate the future use of
attenuated viruses as potential live vaccines. Furthermore, small
molecules directed against signaling antagonists, such as
inhibi-tors of the NS4A-MAVS interaction, may be developed as
anti-DENV therapeutics.
We report that DENV NS4A, an ER membrane-bound
pro-tein, interacts with the mitochondrial protein MAVS. This raises
the question of whether NS4A is a strict membrane-bound protein
or whether it can localize to other intracellular compartments
such as MAMs. A recent study showed that MAM localization of
HCV NS3/4A governs its dual distribution in the ER and
mito-chondria (
46
); therefore, it would be of interest to study whether
MAM could also position DENV NS4A in proximity to the
adap-FIG 7NS4A inhibits VSV-induced IFN. (A) 293T cells were transfected with an empty vector or NS4A-, prM-, or PB1-F2-expressing vectors. Twenty-four hours later, 293T cells were infected with VSV-GFP for 24 h, and the number of GFP-positive cells was scored by flow cytometry. (B) 293T cells transfected with the IFN-reporter together with NS4A, prM, and PB1-F2 (1g/well) were infected with VSV-GFP for 24 h, and cell lysates were then analyzed by a luciferase activity assay. The data are shown as the means⫾SD derived from three repeat experiments. **,P⬍0.01 (Student’sttest). (C) Representative fluorescence micrographs of VSV-GFP in 293T cells transfected with a blank control vector or an NS4A-, prM-, or PB1-F2-expressing vector for 24 h followed by infection with VSV-GFP for 24 h. Mock-infected cells were used as a control. IVA, influenza virus A; DIC, differential interference contrast.
He et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.114.474.63.405.2]tor protein MAVS. Indeed, in our study, DENV NS4A was found
to colocalize and interact with MAVS in the MAM, which may
represent a new cellular localization and function for this
impor-tant DENV nonstructural protein. In addition, in our study, the
transmembrane domain of NS4A could bind the CARD domain
of MAVS. It is possible that the MAM is able to position DENV
NS4A in proximity to MAVS and thus facilitate their interaction
with each other.
Our findings reveal that NS4A binds MAVS through
interact-ing with both the N-terminal and C-terminal domains of MAVS
and subsequently suppresses the binding of endogenous RIG-I.
Notably, the N-terminal CARD-like domain of MAVS was
previ-ously shown to interact with RIG-I and to mediate IFN signaling
(
11
). Additionally, as the C-terminal transmembrane
domain-containing region of MAVS has been found to mediate
oligomer-ization (
47
) and interactions with other adaptor proteins, such as
tumor necrosis factor receptor-associated factor 3 (TRAF3) (
48
)
and TRAF6 (
49
), it is possible that NS4A also disrupts MAVS
oligomerization or the formation of complexes with other adaptor
proteins. Furthermore, it should be noted that another member of
the RLR family, melanoma differentiation-associated gene 5
(MDA5), was previously reported to be a sensor of DENV and to
synergistically mediate type I IFN responses with RIG-I (
50
).
No-tably, MDA5 also has two CARD domains at its N terminus and
transduces its signal through MAVS (
8
), and thus, it may be of
interest to further investigate whether NS4A can disrupt the
inter-action between MAVS and MDA5.
In summary, the present study demonstrates that the adaptor
protein MAVS is a target of DENV NS4A, revealing a novel
func-tion of the DENV NS4A protein in modulating host innate
immu-nity and possibly facilitating DENV infection. The physiological
significance of NS4A in DENV replication and its pathological
role in dengue diseases warrant further investigation into the
po-tential value of this molecule as a therapeutic and/or prevention
target.
ACKNOWLEDGMENTS
This work was supported by the Natural Science Foundation of China (grants 81330058, 81272417, 81501744, and 81571992), the National Mega Project on Major Infectious Disease Prevention (grant 2012ZX10004-213), the Key (Key grant) Project of the Chinese Ministry of Education (grant 311030), the National Science and Technique Major Project (grant 201305017), Guangdong Natural Science Funds for Distin-guished Young Scholar (grant 2014A030306023), and the Guangdong Recruitment Program of Creative Research Groups (grant 2009010058).
FUNDING INFORMATION
This work was funded by National Mega Project on Major Infectious Disease Prevention (2012ZX10004-213). This work was funded by Key Project of Chinese Ministry of Education (311030). This work was funded by National Science and Technique Major Project (201305017). This work was funded by Guangdong Natural Science Funds for Distinguished Young Scholars (2014A030306023). This work was funded by Guangdong Recruitment Program of Creative Research Groups (2009010058). This work was funded by National Natural Science Foundation of China (NSFC) (81330058, 81272417, 81501744, 81571992).
The funders had no role in study design, data collection and interpreta-tion, or the decision to submit the work for publication.
REFERENCES
1.Clyde K, Kyle JL, Harris E.2006. Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J Virol80:11418 –11431.http://dx.doi.org/10.1128/JVI.01257-06. 2.Guzman MG, Halstead SB, Artsob H, Buchy P, Farrar J, Gubler DJ,
Hunsperger E, Kroeger A, Margolis HS, Martinez E, Nathan MB, Pelegrino JL, Simmons C, Yoksan S, Peeling RW. 2010. Dengue: a continuing global threat. Nat Rev Microbiol8:S7–S16.http://dx.doi.org /10.1038/nrmicro2460.
3.Perera R, Kuhn RJ.2008. Structural proteomics of dengue virus. Curr Opin Microbiol11:369 –377.http://dx.doi.org/10.1016/j.mib.2008.06 .004.
4.Sen GC.2001. Viruses and interferons. Annu Rev Microbiol55:255–281.
http://dx.doi.org/10.1146/annurev.micro.55.1.255.
5.Samuel CE.2001. Antiviral actions of interferons. Clin Microbiol Rev
14:778 – 809.http://dx.doi.org/10.1128/CMR.14.4.778-809.2001. 6.Yoneyama M, Fujita T.2009. RNA recognition and signal transduction
by RIG-I-like receptors. Immunol Rev227:54 – 65.http://dx.doi.org/10 .1111/j.1600-065X.2008.00727.x.
7.Seth RB, Sun L, Ea CK, Chen ZJ.2005. Identification and characteriza-tion of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell122:669 – 682.http://dx.doi.org/10.1016/j.cell .2005.08.012.
8.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S.2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol6:981–988.
http://dx.doi.org/10.1038/ni1243.
9.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB.2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell
19:727–740.http://dx.doi.org/10.1016/j.molcel.2005.08.014.
10. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Barten-schlager R, Tschopp J.2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature437:1167– 1172.http://dx.doi.org/10.1038/nature04193.
11. Moore CB, Ting JP.2008. Regulation of mitochondrial antiviral signaling pathways. Immunity28:735–739.http://dx.doi.org/10.1016/j.immuni .2008.05.005.
12. Morrison J, Aguirre S, Fernandez-Sesma A. 2012. Innate immunity evasion by dengue virus. Viruses4:397– 413.http://dx.doi.org/10.3390 /v4030397.
13. Perry ST, Prestwood TR, Lada SM, Benedict CA, Shresta S.2009. Cardif-mediated signaling controls the initial innate response to den-gue virus in vivo. J Virol83:8276 – 8281.http://dx.doi.org/10.1128/JVI .00365-09.
14. Rodriguez-Madoz JR, Belicha-Villanueva A, Bernal-Rubio D, Ashour J, Ayllon J, Fernandez-Sesma A.2010. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. J Virol84:9760 –9774.http://dx.doi .org/10.1128/JVI.01051-10.
15. Diamond MS, Roberts TG, Edgil D, Lu B, Ernst J, Harris E. 2000. Modulation of dengue virus infection in human cells by alpha, beta, and gamma interferons. J Virol74:4957– 4966.http://dx.doi.org/10.1128/JVI .74.11.4957-4966.2000.
16. Diamond MS, Harris E.2001. Interferon inhibits dengue virus infection by preventing translation of viral RNA through a PKR-independent mechanism. Virology289:297–311.http://dx.doi.org/10.1006/viro.2001 .1114.
17. Munoz-Jordan JL, Laurent-Rolle M, Ashour J, Martinez-Sobrido L, Ashok M, Lipkin WI, Garcia-Sastre A.2005. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J Virol79:8004 – 8013.http://dx.doi.org/10.1128/JVI.79.13.8004-8013.2005.
18. Munoz-Jordan JL, Sanchez-Burgos GG, Laurent-Rolle M, Garcia-Sastre A.2003. Inhibition of interferon signaling by dengue virus. Proc Natl Acad Sci U S A100:14333–14338.http://dx.doi.org/10.1073/pnas.2335168100. 19. Ho LJ, Hung LF, Weng CY, Wu WL, Chou P, Lin YL, Chang DM, Tai TY, Lai JH.2005. Dengue virus type 2 antagonizes IFN-alpha but not IFN-gamma antiviral effect via down-regulating Tyk2-STAT signaling in the human dendritic cell. J Immunol174:8163– 8172.http://dx.doi.org/10 .4049/jimmunol.174.12.8163.
20. Jones M, Davidson A, Hibbert L, Gruenwald P, Schlaak J, Ball S, Foster GR, Jacobs M.2005. Dengue virus inhibits alpha interferon signaling by
Targeting MAVS by Dengue Virus NS4A
on November 7, 2019 by guest
http://jvi.asm.org/
reducing STAT2 expression. J Virol79:5414 –5420.http://dx.doi.org/10 .1128/JVI.79.9.5414-5420.2005.
21. Mazzon M, Jones M, Davidson A, Chain B, Jacobs M.2009. Dengue virus NS5 inhibits interferon-alpha signaling by blocking signal trans-ducer and activator of transcription 2 phosphorylation. J Infect Dis200:
1261–1270.http://dx.doi.org/10.1086/605847.
22. Ashour J, Laurent-Rolle M, Shi PY, Garcia-Sastre A. 2009. NS5 of dengue virus mediates STAT2 binding and degradation. J Virol83:5408 – 5418.http://dx.doi.org/10.1128/JVI.02188-08.
23. Yu CY, Chang TH, Liang JJ, Chiang RL, Lee YL, Liao CL, Lin YL.2012. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog8:e1002780. http://dx.doi.org/10.1371/journal .ppat.1002780.
24. Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal-Rubio D, Shabman RS, Simon V, Rodriguez-Madoz JR, Mulder LC, Barber GN, Fernandez-Sesma A.2012. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog 8:e1002934. http://dx.doi.org/10.1371/journal .ppat.1002934.
25. Yamagata T, Nishida J, Tanaka S, Sakai R, Mitani K, Yoshida M, Taniguchi T, Yazaki Y, Hirai H.1996. A novel interferon regulatory factor family transcription factor, ICSAT/Pip/LSIRF, that negatively reg-ulates the activity of interferon-regulated genes. Mol Cell Biol16:1283– 1294.http://dx.doi.org/10.1128/MCB.16.4.1283.
26. Zhou JM, Tang YX, Fang DY, Zhou JJ, Liang Y, Guo HY, Jiang LF.
2006. Secreted expression and purification of dengue 2 virus full-length nonstructural glycoprotein NS1 in Pichia pastoris. Virus Genes33:27–32.
http://dx.doi.org/10.1007/s11262-005-0036-6.
27. Liang Z, Wu S, Li Y, He L, Wu M, Jiang L, Feng L, Zhang P, Huang X.
2011. Activation of Toll-like receptor 3 impairs the dengue virus serotype 2 replication through induction of IFN-beta in cultured hepatoma cells. PLoS One6:e23346.http://dx.doi.org/10.1371/journal.pone.0023346. 28. Lambeth CR, White LJ, Johnston RE, de Silva AM.2005. Flow
cytom-etry-based assay for titrating dengue virus. J Clin Microbiol43:3267–3272.
http://dx.doi.org/10.1128/JCM.43.7.3267-3272.2005.
29. Kochs G, Garcia-Sastre A, Martinez-Sobrido L.2007. Multiple anti-interferon actions of the influenza A virus NS1 protein. J Virol81:7011– 7021.http://dx.doi.org/10.1128/JVI.02581-06.
30. Martinez-Sobrido L, Zuniga EI, Rosario D, Garcia-Sastre A, de la Torre JC.2006. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J Virol
80:9192–9199.http://dx.doi.org/10.1128/JVI.00555-06.
31. Salio M, Ghadbane H, Dushek O, Shepherd D, Cypen J, Gileadi U, Aichinger MC, Napolitani G, Qi X, van der Merwe PA, Wojno J, Veerapen N, Cox LR, Besra GS, Yuan W, Cresswell P, Cerundolo V.
2013. Saposins modulate human invariant natural killer T cells self-reactivity and facilitate lipid exchange with CD1d molecules during anti-gen presentation. Proc Natl Acad Sci U S A110:E4753–E4761.http://dx .doi.org/10.1073/pnas.1310050110.
32. Wu J, Jiang Y, Yang W, He Z, Meng S, Zhang Q, Lin M, Zhang H, Li W, Yang Y, Jia Y, Qian L, Lu D, Cai W, Luo G, Wang Y, Zhu X, Li M. 2012. Dual function of RGD-modified VEGI-192 for breast cancer treatment. Bioconjug Chem23:796 – 804.http://dx.doi.org/10 .1021/bc2006576.
33. Lee S, Tsai YC, Mattera R, Smith WJ, Kostelansky MS, Weissman AM, Bonifacino JS, Hurley JH.2006. Structural basis for ubiquitin recognition and autoubiquitination by Rabex-5. Nat Struct Mol Biol13:264 –271.
http://dx.doi.org/10.1038/nsmb1064.
34. Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P.
2009. Isolation of mitochondria-associated membranes and mitochon-dria from animal tissues and cells. Nat Protoc4:1582–1590.http://dx.doi .org/10.1038/nprot.2009.151.
35. Varga ZT, Ramos I, Hai R, Schmolke M, Garcia-Sastre A, Fernan-dez-Sesma A, Palese P. 2011. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog7:e1002067.http://dx.doi.org/10.1371 /journal.ppat.1002067.
36. Varga ZT, Grant A, Manicassamy B, Palese P.2012. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J Virol86:
8359 – 8366.http://dx.doi.org/10.1128/JVI.01122-12.
37. Vance JE.1990. Phospholipid synthesis in a membrane fraction associ-ated with mitochondria. J Biol Chem265:7248 –7256.
38. Lebiedzinska M, Szabadkai G, Jones AW, Duszynski J, Wieckowski MR.
2009. Interactions between the endoplasmic reticulum, mitochondria, plasma membrane and other subcellular organelles. Int J Biochem Cell Biol41:1805–1816.http://dx.doi.org/10.1016/j.biocel.2009.02.017. 39. Miller S, Kastner S, Krijnse-Locker J, Buhler S, Bartenschlager R.2007.
The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J Biol Chem282:8873– 8882.http://dx.doi.org/10.1074/jbc.M609919200. 40. Loo YM, Gale M, Jr.2011. Immune signaling by RIG-I-like receptors.
Immunity34:680 – 692.http://dx.doi.org/10.1016/j.immuni.2011.05.003. 41. Park MS, Shaw ML, Munoz-Jordan J, Cros JF, Nakaya T, Bouvier N, Palese P, Garcia-Sastre A, Basler CF. 2003. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J Virol77:1501– 1511.http://dx.doi.org/10.1128/JVI.77.2.1501-1511.2003.
42. Charoenthongtrakul S, Gao L, Parvatiyar K, Lee D, Harhaj EW.2013. RING finger protein 11 targets TBK1/IKKi kinases to inhibit antiviral signaling. PLoS One 8:e53717. http://dx.doi.org/10.1371/journal.pone .0053717.
43. Liu SY, Sanchez DJ, Cheng G.2011. New developments in the induction and antiviral effectors of type I interferon. Curr Opin Immunol23:57– 64.
http://dx.doi.org/10.1016/j.coi.2010.11.003.
44. Chang TH, Liao CL, Lin YL.2006. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes Infect8:157–171.http: //dx.doi.org/10.1016/j.micinf.2005.06.014.
45. Rodriguez-Madoz JR, Bernal-Rubio D, Kaminski D, Boyd K, Fernan-dez-Sesma A.2010. Dengue virus inhibits the production of type I inter-feron in primary human dendritic cells. J Virol84:4845– 4850.http://dx .doi.org/10.1128/JVI.02514-09.
46. Horner SM, Liu HM, Park HS, Briley J, Gale M, Jr.2011. Mitochon-drial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A108:14590 –14595.http://dx.doi.org/10.1073/pnas.1110133108. 47. Baril M, Racine ME, Penin F, Lamarre D.2009. MAVS dimer is a crucial
signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. J Virol83:1299 –1311.http://dx.doi.org/10.1128 /JVI.01659-08.
48. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ.2011. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell146:448 – 461.http://dx.doi.org/10.1016/j .cell.2011.06.041.
49. Tang ED, Wang CY. 2009. MAVS self-association mediates antiviral innate immune signaling. J Virol 83:3420 –3428. http://dx.doi.org/10 .1128/JVI.02623-08.
50. Nasirudeen AM, Wong HH, Thien P, Xu S, Lam KP, Liu DX.2011. RIG-I, MDA5 and TLR3 synergistically play an important role in restric-tion of dengue virus infecrestric-tion. PLoS Negl Trop Dis5:e926.http://dx.doi .org/10.1371/journal.pntd.0000926.
He et al.