0022-538X/94/$04.00+0
Copyright © 1994, American Society for Microbiology
Membrane-Binding Domains and Cytopathogenesis
of the
Matrix Protein of Vesicular Stomatitis
Virus
ZHIPING YE, WEI SUN, KALACHAR SURYANARAYANA, PETERJUSTICE, DRUEN ROBINSON,ANDROBERT R. WAGNER*
Departmentof Microbiology and Cancer Center, University of Virginia
Schoolof Medicine, Charlottesville, Virginia 22908
Received 3 June 1994/Accepted 8 August 1994
Themembrane-binding affinityof the matrix(M)proteinof vesicular stomatitis virus (VSV)wasexamined
by comparingthe cellulardistributionofwild-type (wt)virus Mproteinwith that oftemperature-sensitive (ts)
and deletion mutants probed by indirect fluorescent-antibody staining and fractionation of infected or
plasmid-transfected CVI cells. The M-gene mutanttsO23 caused cytopathic rounding of cells infected at
permissivetemperaturebutnotofcellsatthenonpermissivetemperature;wtVSV alsocausesrounding,which
prohibits study of M protein distribution by fluorescent-antibody staining. Little or no M protein can be
detected in theplasmamembrane of cells infected withtsO23atthenonpermissivetemperature, whereas -20% of the M protein colocalized with the membrane fraction of cells infected with tsO23 at the permissive temperature. Cells transfected with a plasmid expressing intact 229-amino-acid wt M protein (M1-229)
exhibited cytopathic cell roundingand actin filament dissolution,whereas cells retained normal polygonal
morphology andactin filamentswhen transfected withplasmids expressingMproteinstruncatedtothe first 74 N-terminal amino acids(M1-74)ordeleted ofthe first 50 amino acids(M51-229)oramino acids 1to50 and
75to 106 (M51-74/107-229). Truncated proteins M1-74 and M51-229were readilydetectable in the plasma
membrane and cytosol of transfected cells as determined by both fluorescent-antibody staining and cell
fractionation,as wasthe plasmid-expressed intactwtMprotein. However, the expressed doubly deletedprotein
M51-74/107-229 could not be detected in plasma membrane by fluorescent-antibody staining or by cell
fractionation, suggesting thepresenceoftwomembrane-binding sites spanning theregion of amino acids1to 50 and amino acids 75to106 ofthe VSV Mprotein.These in vivo datawereconfirmedbyaninvitrobinding
assay inwhich intact M protein and its deletionmutants were reconstituted in high- or low-ionic-strength
buffers with synthetic membranes in the form of sonicated unilammelar vesicles. The results of these
experimentsappeartoconfirm thepresenceoftwomembrane-bindingsitesonthe VSV Mprotein,onebinding
peripherallybyelectrostaticforcesatthehighly charged NH2terminus and the otherstably bindingmembrane
integration of hydrophobic aminoacids and located byahydropathy plotbetweenaminoacids 88and 119.
The matrix(M) protein of vesicularstomatitis virus(VSV)is the mostabundant structural proteinof the virus (25),and it
apparently playsthe central role inassemblyof the virion(22,
26). Amajor portion of theM protein bindsto nascentVSV
ribonucleoprotein (RNP) cores,possibly at the inner surface,
resultingincondensation ofRNPcoresintotight coils (2, 20).
Binding of M protein also results in down-regulation of
transcription by RNP cores (6). The Mprotein is quite basic
and hasaplof -9.1(6), largelybecause ofaplethoraoflysines
and arginines at the amino terminus (23). The positively
chargedamino terminus of the Mprotein appears toprovide
the site forbindingof Mprotein toRNPcores(10, 24).
The affinityinvivo of M proteinfor the plasma membrane
was demonstrated conclusively by the recent finding that at least 10%of allvector-expressedMprotein migratestoplasma
membrane, where it is tightlybound(7).Moreover,expression
of Mproteinaloneresultsinevagination ofM-protein vesicles
fromthe plasma membrane surface (13).Extensive studies of
themembrane-bindingcapacity of the VSV M protein showed
thatwild-type (wt)M protein could bindinvitroto synthetic
lipid vesicles onlyifthe phospholipidscontained acidic
head-groups (27, 31). Affinity labeling and protease digestion
re-*Correspondingauthor.Mailingaddress:Department of
Microbi-ology,School ofMedicine, Box 441, University of Virginia, Charlottes-ville, VA22908. Phone: (804) 924-1948or(804) 924-5111. Fax:(804) 982-1071.Electronicmail address: [email protected].
vealed the presence of a membrane-binding site of the M
proteinatits basic 19 N-terminal amino acids(12). Inanother
study, it was found that M protein missing the first 43
N-terminal amino acids retained somecapacity tobind
phos-pholipid vesicles, suggesting the presence of another
mem-brane-bindingsite distaltothehighly chargedNterminus(21).
Chong and Rose (8) very recently found that a truncated
expressed M protein, missing the first 15 N-terminal amino
acids,retained itscapacitytoassociate withplasmamembrane
of transfected cells but was less tightly bound than plasma
membrane-associated intact Mproteinortruncated Mprotein
devoid of its 14 C-terminal amino acids. The evidence todate thus suggests thepossibilityoftwomembrane-binding siteson
the Mprotein ofVSV.
In an effort to identify more precisely the existence and
location of membrane-binding sites on VSV M protein, we
studied its distribution in CV1 cells infected withwtVSV and
atemperature-sensitive VSVmutantortransfected with
dele-tionmutantsof the Mgene.We alsocomparedthe ionicbinding
stabilitiesonphospholipidvesicles ofwtandmutantMproteins.
The datasuggestthepresenceoftwodistinctmembrane-binding
sites withdifferingaffinitiesontheVSVMprotein.
MATERIALS AND METHODS
Viruses and cell culture. The wt Orsay strain of VSV-Indiana and its group III (M gene) temperature-sensitive
7386
on November 9, 2019 by guest
http://jvi.asm.org/
A.
B.
M
AMINO ACID No. 1 51 74 107 229 + pTM-OM79
pTM (Ml-74) 1_- It
pTM lM5 1-74f107-229)*
pTM(M51-229)
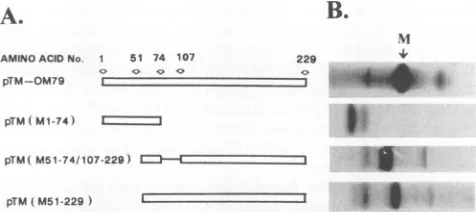
FIG. 1. Stick model (A) of wt anddeletion mutants of the VSV-Indiana cDNA Mgene cloned in vector pTM1 and autoradiographed electropherogram (B) showing migration of35S-labeledwt and deleted M proteins by SDS-PAGE. The numbers in parentheses for the deletion mutants refer to their amino acid sequences, which are also indicated by the arrows above the wt M protein showing relevant amino acid sites foreach deleted protein.
mutant tsO23 were obtained from Anne Flamand of the
UniversitedeParis-Sud, Orsay, France. Virusesweregrownat
31°C on cultures of BHK-21 cells andtitrated by plaque assay
on monolayersof L cells atpermissive (31°C)and restrictive
(39°C) temperatures (24). The vaccinia virus recombinant
(vTF1-6,2) expressing the T7 bacteriophage DNA-dependent
RNApolymerasewas agift ofBernard Moss of the National
Institutes of Health. VSV infection and plasmid transfection/
vTF1-6,2coinfection were performed by using CV1 cells grown
to 95% confluence at 37°C in Dulbecco modified Eagle
medium supplemented with 10% tryptose-phosphate broth,
10% fetal bovine serum, and antibiotics.
Completeand deletedM-geneexpressionvectors.ThewtM
gene used in these studies was a cDNA coding for all 229
amino acids of the Mprotein; itwasobtained from themRNA
of the VSV-Indiana Orsay strain and was cloned in this
laboratory intoavectordesignated pYL-OM79 flankedby the
promoter and terminator sequences for bacteriophage T7
RNApolymerase(14).AtruncatedMgenecoding for the first
74N-terminal aminoacids, designatedpYL(M1-74),was
cre-atedby digestion ofpYL-OM79 with BglII at M-gene
nucle-otide 258 and with
KpnI,
which cleaves at a vector siteimmediately downstream from theMgene;the gapwassealed
with a syntheticoligonucleotide linker, providing an in-frame
stop codon. Atruncated M-gene clone in which the first 50
N-terminal amino acids are deleted, designated
pYL(M51-229), was constructed by PCR using an upstream primer
providing anNcoI cleavage site at an internal in-frame ATG
startcodoncorrespondingtoM-gene nucleotides 249to251;a
downstreamprimerprovidedanEcoRIcleavagesite. Adouble
deletion of theMgene,designated
pYL(M51-74/107-229),
wasalsoconstructedby PCRusinganupstream primerproviding
anNcoIcleavagesiteat aninternal in-frame ATGstartcodon
correspondingtoM-gene nucleotides 249to251 anda
down-streamprimerproviding anEcoRIcleavage siteto delete the
first 50 amino acids andbycleavingatnucleotides 258and358
withBglII todelete amino acids 75 to 106.
Toprovidemaximumexpression ofMproteinsin vitro and
in vivo, the PCR products of pYL(M1-74), pYL(M51-229),
andpYL(M51-74/107-229) wereexcised with NcoI and EcoRI
andinserted between theNcoIandEcoRI sites of the
expres-sionvectorpTM1,kindly provided byBernardMoss(19).The pTM1 vector contains theT7bacteriophage polymerase
pro-moter andterminator sequences, aswell asthe
cap-indepen-dent translation property. Figure 1 shows stick models for the
M-gene deletion plasmids pTM(M1-74),
pTM(M51-74/107-229), and pTM(M51-229), as well as the complete M gene
cloned in pTM1 and designated pTM-OM79 (Fig. 1A). Also
shown bysodium dodecyl sulfate- olyacrylamide gel
electro-phoresis (SDS-PAGE) are the [3 S]methionine-labeled
pro-teins expressed by these four plasmids in a TnT in vitro
coupledtranscription-translationsystem(Fig. 1B). These
35S-labeled complete and truncated M proteinswere purifiedby
affinity chromatography on a column of Sepharose coupled
withmonospecificpolyclonal antibody made in rabbits
immu-nized withpurifiedMprotein.
Immunofluorescence studies. CV1cellsweregrownonglass
coverslipsfor 2 dayspriorto infection withwt orts023 virus
(multiplicity of infection of -5 PFU per cell) or to plasmid
transfection. Infected or transfected cellswere incubated for
various periods at temperatures chosen to provide optimal
permissive (31°C) ornonpermissive (39°C) conditions for the
mutant tsO23. For transient expression of mutant or wt M
proteinscloned in plasmids, theCV1 cellswere first infected
with the vaccinia virusrecombinantvTF1-6,2expressingthe T7
polymerase (multiplicity of infection of 5 PFU per cell) and
then transfected with plasmids containing M-gene mutants
(0.01 mg) in the presence ofLipofectin. After incubation for
various periods, the infected or transfected CV1 cells were
washed incold phosphate-buffered saline (PBS), fixed for 20
min in 3% formaldehyde, andpermeabilized by exposure to
0.4%Triton X-100 for 3 min.Nonspecific stainingwasblocked
by incubation of the cells for 30 min in 3% bovine serum
albumin in PBS. The infected or transfected cellswere then
incubated at room temperaturefor 40 min with rabbit anti-M
proteinserum(immunoglobulinG[IgG])at aconcentration of
0.005
mg/ml
in PBS and withrhodamine-phalloidin
(to
stainactin
filaments).
Eachcoverslipwasthenflooded with donkeyanti-rabbit IgG conjugated with fluorescein isothiocyanate
(FITC) and incubated for 40 min at room temperature.
Washed coverslips were then mounted in 90%
glycerol
and10% PBS in 3,4,5-trihydroxybenzoic acid N-propylester to
prevent photobleaching. Cellular distribution of
immunofluo-rescence was determined with atransmission microscope
un-deranepifluorescentUVlightsource.Cellularmorphologyof
the same field was examined by using phase-contrast
micro-graphs underaregularlightsource.
Fractionation of infected and transfected cells.
Subconflu-ent monolayers of CV1 cells in 60-mm-diameter plates were
first infected with VSV or the T7 polymerase recombinant
vTF1-6,2 by30 minofadsorptionatamultiplicityof infection
of 5 PFU per cell. The
vTF1-6,2-infected
cells were thentransfected with 10 ,ug ofM-generecombinantplasmid cDNA
assistedbyLipofectinfor 2 hat37°C.After
being
washed withPBS, the cells were incubated for 1 h at 37°C in 2 ml of
methionine-free minimum essential medium with 5% fetal
bovine serum and 0.05 mCi of
[35S]methionine.
Thelabeling
medium was then replaced with 2 ml of minimum essential
medium containing 5% fetal bovine serum and 5 mM
unla-beled methionine. After further
incubation,
the35S-labeled
cells were washed with PBS and harvested
by scraping
themonolayers in homogenization buffer
(HB;
10 mM Tris-HCl[pH7.4],10% sucrose,1 mMEDTA,1,ugof
aprotinin
perml).
Cells were disrupted by 20 strokes in a
tight-fitting
Douncehomogenizer (Thomas; 0.1-nm
clearance)
cooledby
ice,
and the lysateswereclarified bycentrifugation
at2,000
xg for 5minat4°C.Theclarifiedlysateswerethen
layered
on a4.5-ml10 to 70% continuous sucrose
gradient
forcentrifugation
at100,000 xg for 20hat
4°C.
The fractionswerecollected from thebottom,diluted withequal
volumesof2xNET-gel
buffer(50mMTris-HCl
[pH
7.5],
150mMNaCl,
0.1% NonidetP-40,
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.57.295.80.186.2]1 mM EDTA, 0.25%
gelatin,
0.02% sodiumazide),
andimmunoprecipitated
with rabbit anti-M protein polyclonalserum
(1:500
dilution ofserum).
The35S-labeledimmunopre-cipitated proteins
in each fraction wereanalyzed by
SDS-PAGE and scored
by integrated densitometry.
Invitro reconstitution ofMproteinswithsonicated vesicles.
Complete
anddeleted VSV Mproteins
labeled with[35S]me-thionine were
synthesized
in vitro fromM-generecombinantpTM1
plasmids by using
thePromegaTnTcoupledtranscrip-tion-translation kit driven
by phage
T7polymerase
andtrans-lated ina
reticulocyte lysate.
Theefficiency
of[35S]methionine
incorporation
intonewly
translatedproteins
was22%,
withbackground incorporation
of 0.2%. All translated Mproteins
were
purified by affinity chromatography
on a column ofSepharose
conjugated
withpolyclonal antibody specific
for Mprotein.
The sonicated vesicles wereprepared
as previouslydescribed
(29).
Inshort,
500 nmolofdipalmitoylphosphatidyl-choline
(DPPC)
wasmixed with 500 nmol ofdipalmitoylphos-phatidylglycerol
(DPPG);
this 1:1 molar ratio ofDPPC/DPPGwasdried under
nitrogen
andlyophilized overnight.
Thelipid
samples
werethensuspended
in2ml of 10mMTricine buffer(pH 7.4) containing
nosalt,
0.15MNaCl,
or0.65MNaCl andsonicated at
42°C
for 3 to 5 min undernitrogen
with aHeat-Systems
W-350 sonicator. Multilamellar vesicles wereremoved
by centrifuging
thesonicatedlipid samples
at150,000
xgfor 3 h. Theresultingsmallunilamellar vesicles(about25
nmol)
weremixed withwt ortruncated35S-labeled
Mproteins(150,000 cpm)
andwerethendialyzed
for 24 hagainst 10 mMTricine
containing
the desired NaClconcentrations. Thevesi-cles reconstituted with M
protein
were thensubjected
tobuoyant-density (isopycnic) centrifugation
for 16 h at200,000
x g at 46°C in a 0 to 30% sucrose gradient as previously
described
(27). Samples (0.4
mleach)
wereanalyzed
forbuoyant
density byrefractometry,
and thegradient
distribution of35S-M
proteins was determinedby
SDS-PAGE andautora-diography
andintegrated by densitometry.
RESULTS
Cellular distribution oftsO23Mprotein atpermissiveand
restrictive temperatures.Ithasbeen known forsometime that
the VSV M
protein
lines the inner surface of the virionenvelope
and bindsto the inner surface of the plasmamem-brane,
aswell as the RNP cores, in VSV-infected cells(26).
Chong
andRose(7)
have shownthat,
inthe absence of other VSVproteins,
90% of theplasmid-expressed
Mprotein
isdistributed in the
cytosol
and 10% is associated with theplasma
membranes of transfected cells. It was of interest todetermine whether at
nonpermissive
temperatures the VSVM-gene
conditionalmutanttsO23,
which is defective in virionassembly, synthesizes
Mprotein
restricted inbinding
toplasma
membrane. Because of the
cytopathic
effect of cell roundingcaused
by
Mprotein synthesized
incells infectedwithwtVSV or itsM-gene
mutant tsG33 at permissive temperatures (5),fluorescent-antibody
labeling studies were likely to be oflimited use in determining the distribution of M protein in
infectedcells. For this reason, cellfractionation studies were
also
performed.
It was also of interest to compare the cellrounding
inducedbytsO23 withthatinducedbythe differentM-gene
mutanttsG33 testedpreviously (5).Inthese
experiments,
CV1 cellswereinfectedwith wtVSVortsO23andincubated for4 hat
310
(permissive
temperature)or
39°C (restrictive temperature).
As described in Materials andMethods,
infected CV1 cells grown on coverslips wereexposed
tomonospecific
rabbit anti-Mserumandlabeled withFITC-conjugated
donkeyanti-rabbit IgG forexamination byfluorescence and phase microscopy. In the secondexperiment,
semiconfluentmonolayers of CV1 cells similarly infected with
tsO23 and labeled with [35S]methionine were incubatedat 31
or 39°C for 4 h and disrupted by Dounce homogenization,
clarified, and fractionated by isopycnic centrifugation on a 10
to70% continuous sucrose gradient, and the distribution of M
protein in each fractionwasdeterminedby
immunoprecipita-tion andanalyzed by SDS-PAGE and integrated densitometry
after autoradiography.
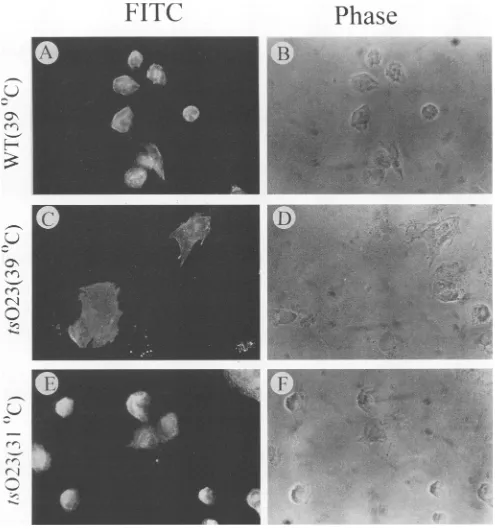
Figure 2 shows the fluorescent staining and phase micro-graphs of representative fields of CV1 cells infected at 39°C with wt VSV and at 31 or 39°C with tsO23. Clearly, the cells
were rounded and shrunken after infection with wtvirus at
39°C (Fig. 2AandB) orwith ts023at 31°C (Fig. 2E and F).
The M protein was uniformly distributed and could not be
localizedtoanyregion of the cells infected with wt virus (Fig.
2A) orwith tsO23 at 31°C (Fig. 2E). In sharp contrast, CV1
cells infected withtsO23 at39°C retained their large size and
typical polygonal morphology when examined by fluorescence
microscopy (Fig. 2C) orby phase microscopy (Fig. 2D). The
tsO23Mprotein appearedtobewidely distributed throughout
the cytoplasm of cells infected at 39°C, with no particular
localization to the surface membrane (Fig. 2C). Quite
obvi-ously,the membranelocalization,if any, incells infected with
wtVSVortsO23at31°Ccannotbe determined because of the
cytopathic effect. Of some interest is the finding that tsO23
causes the samecell roundingat permissivebut not
nonper-missive temperatures asdoes another M-gene mutant, tsG33
(5).
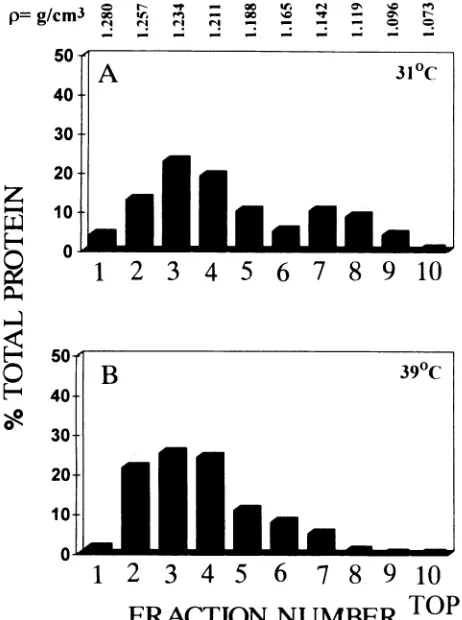
Figure 3 shows the distribution of 35S-labeled M protein
synthesized in CV1 cells infected at 31°C (Fig. 3A) or 39°C
(Fig. 3B)withtsO23 afterdisruption, fractionation by isopycnic
centrifugation, immunoprecipitation, SDS-PAGE,
autoradiog-raphy, and integrated densitometry. As noted, -75% of theM
protein synthesized at 31°C banded at a mean density of
-1.234
g/cm3
and-25% of the M protein banded at a peak of-1.130 g/cm3 (Fig. 3A). The radioactivity at high density is
consistent with the presence of freeMprotein in the cytoplasm
orofM protein in association with nascent RNP cores. The
lowerdensity peak at 1.142g/cm3indicates the presence of M
protein associated with plasma membrane, which bands at this
densityin sucha sucrosegradient(1).Bycomparison, virtually
all the ts023Mprotein synthesizedat39°C banded at a density
of 1.234 g/cm3, except for a trailing decreasing amount of
radioactivitynearthe top of thegradient (Fig. 3B).
The question arises whether the tsO23 M protein fails to
bind to plasma membrane at39°C because it can be
seques-tered on the perinuclear membrane or cytoskeleton at the
nonpermissive temperature. This is particularly relevant
be-causeof thestudies of Onoetal.(21a), who found that theM proteinof thephenotypically similar VSVmutanttsG33 forms
perinuclear aggregates at nonpermissive temperatures but is
diffusely distributedin the cytoplasm at permissive
tempera-tures.Therefore, weanalyzed by the same method(21a) the
distribution of M protein in the nuclei, cytoskeletons, and
cytoplasm of CV1 cells infected with tsO23 at31 and39°C in
the presence of
[35S]methionine.
Briefly, tsO23-infected CV1cells washed at 6 h postinfection at 31 and 39°C were
sus-pended in a HEPES
(N-2-hydroxyethylpiperazine-N'-2-eth-anesulfonicacid)buffer anddisrupted by adding 0.5% Triton
X-100 for 30 s. Aftercentrifugation of the disrupted cells at
1,000 xgfor 5 min, the soluble supernatant fluid was removed
foranalysis. Thepelleted materialwas separated into nuclei andcytoskeletonby homogenization in lysis buffer containing 1%Tween 20 and0.5% deoxycholate, and then it was
centri-fugedat1,000xg for5min.The
35S-labeled
Mprotein in eachon November 9, 2019 by guest
http://jvi.asm.org/
FITC
Phase
41--5 .:A
..
ip
..
FIG. 2. Indirectfluorescent-antibody stainingof Mproteinandphase microscopyof CV1cells infected4hpreviouslywithwtVSV-Indianaat 39°C (AandB)orwith itsM-genemutantts023atthenonpermissive temperatureof39°C (CandD)orthepermissivetemperatureof31°C (E andF).As described in Materials andMethods,infected CV1 cellswerefixed, permeabilized, exposedtorabbit anti-Mmonospecific polyclonal antibody,andlabeled withFITC-conjugated donkeyanti-rabbitIgG. Photomicrographs showcells in thesamefieldsexaminedbyfluorescence
microscopyandphase microscopyatamagnificationof x472.
fraction was immunoprecipitated and quantitated by
SDS-PAGE, autoradiography, and integrated densitometry. The
results showed that the distribution oftsO23 M protein syn-thesized in CV1 cells at the permissive temperature was as
follows: 49% in the cytosol, 20% associated with nuclei, and
31% associatedwith thecytoskeletonfraction. By comparison,
tsO23Mprotein synthesizedatthenonpermissivetemperature
was distributed in CV1 cells as follows: 30% in the cytosol,
40% in the nuclear fraction, and 30% in the cytoskeletal
compartment (datanot shown). These data indicate that the
sequestrationoftsO23Mproteininperinuclearorcytoskeletal
fractions is not responsible for its failure to bind to plasma
4*
"WI'
&
U
Al-rn]
0-
I-Cq
CD
#-,
rp, s-~
z--Z
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.56.551.67.596.2]- 0 tI, e-4 O %Q4 f
- N
r4 0:.; 0
50 e
A
3'V
40
-30 -20
10
i.
0-H0-
1
2
3
4
5
6 7
8 9
H
0-0
H-Oc
10
50-1
23
4 5 67
8
910
FRACTION
NUMBER
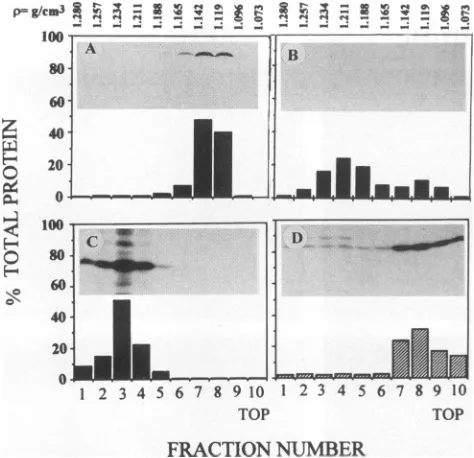
TOPFIG. 3. Distribution of Mproteinincytoplasmand membrane of fractionated CV1 cells infected with VSV mutant tsO23 at the
permissivetemperature of31°C
(A)
orthenonpermissivetemperatureof39°C(B).Cells labeled with[
'S]methionine
wereharvestedat4h after infection anddisrupted byDouncehomogenization.Asdescribed in Materials andMethods, disruptedcellswerefractionatedbyisopyc-niccentrifugationina10to70%sucrosegradient.The35S-labeledM
proteinineach of thetenfractionswasimmunoprecipitated by adding
rabbit anti-M serum in the presence of 0.4% Triton X-100 and
subjected toSDS-PAGE and autoradiography. The relative
propor-tionsof35S-Mproteinineach fractionweredeterminedby integrated
densitometry,and thebuoyant density (p)wasdeterminedby
refrac-tometry.
membraneatthe
nonpermissive
temperature, since almostasmuch tsO23 M
protein
was soluble in the cytoplasm andavailable formembrane
binding
atthepermissive
temperatureas atthe
nonpermissive
temperature.These data indicate that mutant tsO23 M
protein
synthe-sized in cells at the restrictive temperature binds only
mini-mally,
ifatall,
toplasma membrane,
suggestingthatthere isaconditional defectin membrane association of the mutant M
protein.
The distribution of75%cytoplasmic
and 25%mem-brane-associatedtsO23 M protein atthe permissive
tempera-ture is not
significantly
different from the 10% binding toplasma
membrane ofvector-expressed wt Mprotein reportedby Chong
and Rose(7).
Cellular distribution of plasmid-expressed complete and
truncated Mproteins.Itwasof interesttodetermine whether
the intactwtM
protein
andsomeof itsdeletionmutants wereassociatedwith cell membranein the absence of other
intra-cellular VSV
proteins. Therefore,
CV1 cells were transfectedwith
pTM1
recombinantsexpressingtheentirewtMproteinorits deletion mutants depicted in Fig. 1 and coinfected with
vTF1-6,2 vaccinia virus expressing T7 polymerase. In one
experiment, CV1 cellsgrown on coverslipswere fixed at 16 h
posttransfection and exposed to polyclonal rabbit anti-M
se-rum followedby FITC-conjugated donkey anti-rabbit IgG or
by rhodamine-phalloidin tostain actin filaments. Theshape of
these same fluorescently labeled cellswasalso determinedby
phase microscopy. In the second series of experiments,
plas-mid-transfected CV1 cells expressing intact M protein or its
deletion mutants werefractionated and the distribution of M
proteins between cytosol and membrane was determined by
equilibrium (isopycnic) centrifugation as described in
Materi-als andMethods.
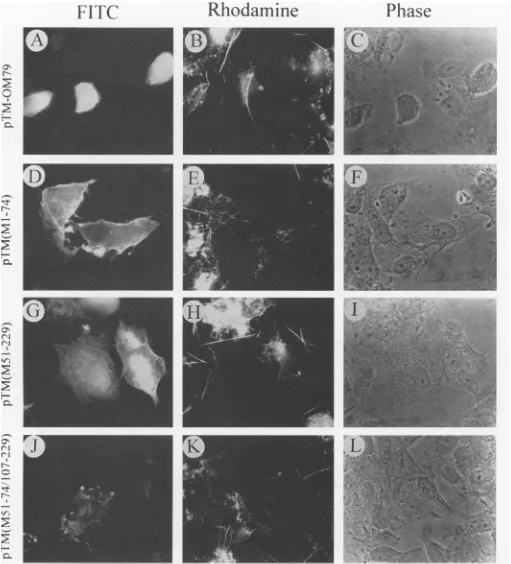
Figure 4 shows the cellular distribution of intact Mprotein
and its deletionmutants, the presence of actin filaments, and
the shape of the cells at 16 h after transfection with
pTM-OM79, pTM(M1-74), pTM(M51-229), or
pTM(M51-74/107-229). As indicated in Fig. 4A through C, expression of the
entirewt Mproteinresulted in markedroundingoftransfected
CVIcells,apaucityof actinfilaments,and uniformfluorescent
staining of M protein, in a manner quite similar to that
observed forCV1 cells infected with wt VSV or withts023at
31°C(Fig. 2).Insharp contrast, CV1 cells transfectedwiththe
threeM-genedeletion mutants retained theirpolygonalshape
and actin filaments as judged by fluorescence and phase microscopy despite ample expression of FITC-stainable M
protein. The distribution of FITC-stainableMprotein and the
presenceof actin filamentsreactivewithrhodamine-phalloidin
are clearly visible in cells transfected with all three deletion
mutants.The M protein truncatedtothe first 74 amino acids,
expressed in cells transfected with pTM(M1-74),was clearly
found at the cellsurface, presumablyattheplasma membrane,
as well as in the cytoplasm (Fig. 4D). Similarly, CV1 cells
transfected with pTM(M51-229), in which the first 50 amino
acidsweredeleted, expressed this truncated Mprotein, which
is distributedinthecytoplasm and partially in the region of the
surfaceplasma membraneaswellasthe nucleus(Fig. 4G). By
comparison, deletion of amino acids 1 to 50 and 75 to 106 in
pTM(M51-74/107-229) resulted in the synthesis of a doubly
deleted Mprotein detectable onlyas punctateclusters in the
cytoplasm, andnotthesurfacemembrane, of transfectedCV1
cells(Fig. 4J). These findings indicate that the entireMprotein
is required for causing a cytopathic effect resulting in cell
rounding and dissolution of actin filaments. In addition, two
deletions arerequired to prevent the association of M protein
withplasma membrane, suggesting the presence oftwo
mem-brane-binding sites on M protein located in the regions of
amino acids 1 to 50 and75 to 106.
Figure 5 provides additional data concerning the membrane
and cytosol distribution of 35S-labeledMprotein expressedin
CV1 cells transfectedwith pTM-OM79 (expressingtheentire
wt M protein), pTM(M1-74), the double-deletion mutant
pTM(M51-74/107-229),andpTM(M51-229), as determined by
cellfractionation, equilibrium density centrifugation,and
SDS-PAGE. As noted, -90% ofthe wt M protein expressed by
pTM-OM79 banded at a mean density of -1.131 g/cm3,
equivalent to the density expected for membrane (Fig. 5A).
Much lower yields of M protein truncated to the first 74
N-terminal amino acids were recoverable from
pTM(M1-74)-transfected CV1 cells, but cell fractionation revealed that
-70%of thisproteinwaspresent incytosol at a mean density of1.211g/cm3 and-30%waspresentin aputative membrane
fraction at a density of 1.119 g/cm3 (Fig. SB). The doubly
deleted M protein expressed by pTM(51-74/107-229)
ex-pressed in CV1 cells fractionated by isopycnic centrifugation
banded entirely at a mean density of 1.234 g/cm3, i.e., at a
p=g/cm3 X N en
*- *;
.r
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.68.299.73.383.2]FITC
Rhodamine
r
r-_2
0
1
r-1
C.
C"l
H
Phase
I
CL
FIG. 4. Indirectimmunofluorescent-antibodystainingofMprotein,actinfilamentlabeling,andphase microscopyof CV1cells transfected with plasmids expressingtheentire VSV M gene,
pTM-0M79
(A through C),orplasmids expressingdeletedMgenes,pTM(Ml-74) (D through F), pTM(M51-229) (G throughI),
orpTM(M51-74/107-229) (J through L).Asdescribed in Materials andMethods,
CV1 cellswereinfected withT7polymerase-expressingvTF1-6,2and transfectedwith recombinantM-geneplasmidsinthepresenceofLipofectin.After incubationat370Cfor16 h aftertransfection,cellswerefixed, permeabilizedwith 0.4% TritonX-100,exposedtorabbit anti-Mserum,and stainedwithFITC-conjugated donkey anti-rabbitIgGandrhodamine-phalloidin.Thephotomicrographsshowrepresentativefields of thesamecellsexamined for distribution of Mprotein byfluorescein labeling (A,D,G, andJ), for actinfilamentsbyrhodamine fluorescence (B, E, H, andK), orforshapebyphase microscopy (C, F,I, andL). Magnification, X458.
I
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.53.564.80.644.2]p=gcm3 F zl _ fX0 Il " -, = t - i o
._______.___________
EH C
0
0
H0
80
-
60-40
-20
0-A
IWt C: 0., ...
60
40
20
ai n M I.
1 2 3 4 5 6 7 8 9T10 2 3 4 5 6 7 8 9 10
TOP TOP
FRACTION NUMBER
FIG. 5. Distribution of M protein between the cytoplasm and plasma membrane of CV1cells transfected withpTM-OM79
express-ing the entire M protein (A), pTM(Ml-74) expressing M protein truncated to the first 74 amino acids (B), pTM(M51-74/107-229) expressingMproteindeleted of amino acids1to50 and 75to106(C),
orpTM(M51-229) expressingMproteindeleted of the first 50 amino acids (D).Asdescribed in Materials andMethods, CV1 cells previ-ously infected with vTF1-6,2 were transfected with each plasmid, labeled with [35S]methionine,anddisrupted byDounce homogeniza-tionafter incubation for 16 h. After removal ofdebrisandnucleiby low-speed centrifugation, the disrupted cells were fractionated by isopycnic centrifugation on a 10to 70% sucrose gradient. The 35S-labeledMprotein in each of the 10 gradientfractionswas immuno-precipitatedwith rabbit anti-Mserumand analyzed bySDS-PAGE. Autoradiographsof Mproteins (shownabove bargraphs)werethen integrated by densitometry,andbuoyant density (p)wasdeterminedby refractometry. The results are plotted as the percentage of 35S-M proteinineachgradientfraction.
density expected for cytosol devoid of membrane; virtually
noneof the Mprotein missingamino acids 1 to50 and 75to 106 could be detected at a density expected for plasma
membrane(Fig. SC).In markedcontrast,when CV1 cellswere
fractionatedby equilibrium density centrifugationafter trans-fection withpTM(MS1-2229) expressingMproteinfrom which
only the first 50 amino acids were deleted, -80% of the truncated Mproteinbandedatameandensityof -1.119g/cm3
in presumptive association with cell plasma membrane (Fig.
SD).
These findings by cell fractionation appear to confirm the data obtainedby fluorescent-antibody staining, indicating the presence of membrane-binding sites on M protein in the
regionof the first 74 aminoacids aswell as aregion distalto
amino acid 51. The failure of the M protein expressed by
pTM(MS1-74/107-229) to bind to low-density cell fractions
indicates the presence oftwo membrane-binding sites on M
protein located between amino acids 1 to 50 and between
amino acids75 to106.
It is incumbent upon us to comment on why M protein
expressed by pTM-OM79 and by pTM(MS1-229) should be
foundsoabundantlyatisopycnicdensities consistent with that
ofmembranes,whereasthe Mprotein expressed during
infec-tion with wt VSVor ts023 at the permissive temperature is localizedinisopycnicfractionated cellsmoreabundantlyat the
densityofcytosolthan at that of membrane(Fig. 3).Apartial
explanation may be that M protein synthesized during VSV
infection is predominantly associated with nucleocapsids,
which band at high densities. However, only 10% of the M
protein expressedinHeLa cellsbyarecombinantplasmidwas
found to colocalize with plasma membrane at the 65%-10% interface of a discontinuous isopycnic sucrose gradient,
whereas -90% ofthe expressed Mproteinwasfound at the 80%-65% sucrose interface thoughtto be in acytosolic
frac-tion (7). Apossible explanation for this discrepancy is that
expressionof theM-gene construct inHeLa cellswasfor 6 h
(7), whereas in our studies the M-gene constructswere
ex-pressed for 16 h, ostensibly providing a longer time for M
proteinstomigratefrom thecytosoltotheplasmamembrane.
Whenwe repeated this experiment by the method ofChong
and Rose (7, 8), using isopycnic flotation centrifugation in a
discontinuous 80%-65%-10% sucrose gradient for fraction-ation of cells transfected withpTM-OM79,wefound that21% of the expressed M protein colocalized at the 65%-10% sucrose interface (presumably with plasma membrane) and 79% was at the 80%-65% (presumably cytosolic) sucrose interface (data notshown). The same experimentwith CV1 cells transfected withpTM(M51-229) revealedthatonly38% of the truncated M protein colocalized with cytosol at the 80%-65% sucroseinterface,whereas 62% of the truncatedM
protein was found in the presumptive membrane-associated
fraction at the 65%-10% sucrose interface (datanotshown).
Thequestionarisesastothereliabilityof cell fractionationby
continuous sucrose isopycnic gradient centrifugation
com-paredwith that ofadiscontinuous sucrose gradient in which
buoyant densitycannot be measured. Regardless, the doubly
deleted M protein expressed by pTM(M51-74/107-229) is
clearly notmembrane associated (Fig. 5C).
Vesicle binding affinity ofintact and deleted M proteins.
Thepreceding experiments suggestthepresenceof twoin vivo
membrane-bindingsitesonthe MproteinofVSV,onelocated
within the first 50amino acids and the other between amino acids 75 and 106. The existence of a similar N-terminal
membrane-binding site on M protein was recently
demon-stratedby Chong and Rose (8), who also demonstrated that
stabilityof membranebindingunderhigh-saltconditionswas
attributable to the C-terminalregionof theMprotein.Earlier studies from our laboratory had revealed that wt VSV M
proteinbinds in vitro tosynthetic phospholipidvesiclesonlyif
they contain acidic headgroups that recognize the basic M
protein (27, 31). It seemed logical therefore to determine
whether the two putative membrane-bindingsiteson M pro-tein contribute to itsbinding affinityfor acidicphospholipids,
aswellaswhether ionicand/orhydrophobicbondscontributed tothe twoputative bindingsites.
In these experimentsunilamellar vesicles containing equal
concentrations of DPPC and DPPG (DPPC/DPPGvesicles)
were prepared bysonication and purified to homogeneityas
previouslydescribed(27, 29).Mproteinslabeled with
[35S]me-thionine were prepared in vitro with the TnT coupled
tran-scription-translation system by using as templates the T7
polymerase-driven plasmidspTM-OM79,expressingthe entire
wt M protein, and pTM(M1-74), pTM(M51-229), and pTM
(M51-74/107-229),each ofwhich expresses the relevant trun-cated M protein (Fig. 1; see Materials and Methods). Soni-cated DPPC/DPPG unilamellar vesicleswere incubated with 35S-labeled entireortruncated Mproteinsin buffers
contain-ing no salt, 0.15 M NaCl, or 0.65 M NaCl. As described in
MaterialsandMethods,theinteractingvesicles and Mproteins
B
. _.I _
--ONNIA-M.6
.
on November 9, 2019 by guest
http://jvi.asm.org/
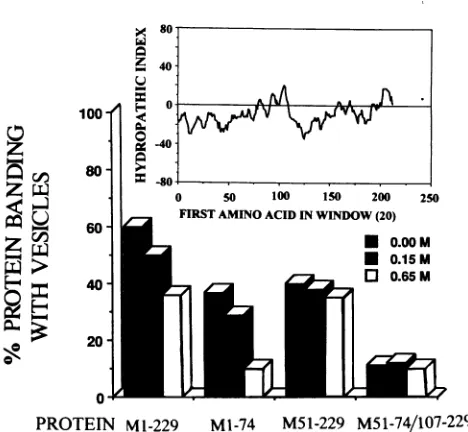
[image:7.612.67.304.72.301.2]I 80
i 40
-40
-80
0 50 100 150 200 25
2 FIRSTAMINOACID INWINDOW(20)
[image:8.612.58.292.59.275.2]
0-PROTEIN
Mrl-229
Ml-74 M51-229M51-74/107-229
FIG. 6. Salt-dependent affinityfor invitrobindingof Mprotein and its deletionmutants tosonicated unilamellar phospholipidvesicles and hydropathy plotof wt VSV Mprotein.Complete M protein containing all 229 amino acids (M1-229) and deletion mutants of M protein truncatedtothe first 74 amino acids(M1-74), deleted of thefirst50 amino acids(M51-229),ordeletedof aminoacids1 to50 and 75 to 106 (M51-74/107-229) were synthesized in vitro and labeled with [35S]
methionine in the TnT coupled transcription-translation system. Unilamellar vesicleswere prepared bysonication ofequimolar con-centrations of DPPC and DPPG. Mixtures of liposomes and 35S-labeledcompleteortruncated Mproteinswerereactedat42°Cfor 24 hduring dialysis againstTricine buffer withnosaltor0.15or0.65 M NaCl. Vesicles bandedby buoyant-density centrifugationina contin-uous0to30%sucrosegradientweretested fortheirconcentrations of bound35S-Mprotein by SDS-PAGE, autoradiography,andintegrated densitometry.The percentage of Mprotein boundtovesiclesateach saltconcentrationwascalculatedfromthetotal
35S-protein
throughout thegradient.The inset shows thehydropathyindex ofwtVSV-Indiana Mprotein calculated by movingsegments of 20 amino acid residues accordingtothe valuesassigned by Kyte and Doolittle(11).were subjected to isopycnic centrifugation in a 0 to 30%
continuous sucrose gradient and the percentage of 35S-M
protein banding with lipid vesicles was measured by
SDS-PAGE, autoradiography, and integrated densitometry. Figure 6 shows bar graphs of the percentages of 35S-M protein associated withDPPC/DPPGvesicles after interaction
in buffercontainingnoNaClor0.15or0.65 M NaCl followed
by isopycnic centrifugation. Also shown inFig.6forreference isahydropathy plotof the entirewtMprotein.Asindicated,
in areaction mixture containingnosalt, 60% of the whole M
proteinboundtoDPPC/DPPG vesicles, comparedwith37% of theMproteintruncatedtothefirst 74 aminoacids, 40%of the
Mprotein extendingfrom amino acid 51to229,andonly 11%
of the double-deletionM51-74/107-229 protein; 10 to 12% of
thebindingcanbe assumedtorepresentbackground
nonspe-cific binding of M protein. We interpret this last result as
indicating that therewasessentially nobinding of themutant
Mproteindeletedin tworegions(fromamino acids1 to50and
75 to 106) to DPPC/DPPG vesicles; these data appear to
support thepreceding studies indicating the complete loss of
membrane binding in vivo of this doubly deleted mutant M
protein. When thewt and truncated M proteinswere
recon-stituted with DPPC/DPPG vesicles at physiological salt
con-centrations
(0.15
MNaCl),
theirbinding
affinities decreasedslightly:therewas a10%decrease for the complete M protein
(M1-229),an8%decrease for truncatedproteinM1-74,and an
insignificant 2% decrease for truncated protein M5 1-229.
Increasing the salt concentration in the vesicle-M protein
binding mediumto0.65MNaClresulted in decreasedbinding
of the entireMprotein(M1-229),from 50to60%to29%,and
of theMproteintruncatedtothe first74amino acids(M1-74),
from 29to37%to10%, essentially to the background level for
thisprocedure. Incontrast, thebinding reaction performed at
0.65MNaCl reduced thebindingtovesicles of theMprotein
from which the first 50 amino acidswere deleted (M51-229)
from alevel of 38to 40% to 35%, an insignificant decline in
binding affinity at a high salt concentration. Quite similar
binding affinities were observed when 14C-labeled wt and
mutant Mproteinswereinteractedwithsonicated
3H-DPPC/
DPPG unilammelar vesicles in the presence of no NaCl or
0.15 or0.65 MNaCl before isopycnic centrifugation. Allbut
the double mutant
14C-M51-74/107-229
colocalized with3H-DPPC/DPPGvesiclesatlow saltconcentrations, and
14C-M1-74
showed no affinityforbinding to
3H-DPPC/DPPG
vesicles inthe presence of 0.65M NaCl(datanotshown).
These data indicate that theMproteintruncated tothefirst
74 aminoacids(M1-74)bindstoDPPC/DPPGvesiclesbyionic
bonds,whereas theMproteininwhichonlythe first 50 amino
acids are deleted (M51-229) appears to bind DPPC/DPPG
vesiclesbynonionicbonds, since it isnotsignificantly impeded
inbindingin the presence of 0.65 MNaCl. Inthecaseof the
entire M protein (M1-229), both ionic and nonionic binding
appear tobeoperative,withthe greaterbinding force
appar-ently being nonionic in character. This
finding
is consistentwith thestudy reported by ChongandRose(8),who found that
membrane-prebound M protein lacking the first 15 amino
acidswasonly slightlymoredissociableby highsalt
concentra-tions than was the membrane-prebound intact M protein,
indicating the presence of a carboxy-distal nonionic mem-brane-binding site.
It is well known that the N-terminal region of VSV M
proteinis highly chargedand rich in basiclysine andarginine
residues(23, 24),thusexplainingitsbindingaffinityforvesicles
containing phospholipids with acidic headgroups (27, 31). Figure6
(inset)
showsahydropathy plot,createdaccordingtothe method ofKyte and Doolittle (11), indicatingtheregions
of the wt VSV M protein possessing
significant
stretches ofhydrophobic amino acids. As indicated, the region between
amino acids 88 and 119canbe consideredtobe
hydrophobic.
This couldexplain whythe deletion ofamino acids 75to106 in
thisregion (alongwith amino acids1 to
50)
results inalossofcapacity tobind cell membrane and
lipid
vesicles. This could alsoexplain why bindingatthisregionisnotprevented by high
salt concentrationsas is
binding
atthe otherputative binding
site spanning the first 50 amino
acids,
whichquite
obviously
binds to membranes through electrostatic forces. The other
hydrophobic region at the extreme C-terminal end
(amino
acids 202 to
208)
of theMprotein apparently
is notlikely
toplay a role in stable membrane association; deletion of 14
C-terminal amino acids did not
disrupt binding
of thistrun-catedM
protein
(8),
and it would notbeexpected
todo so.Finally,we set out todetermine thelevels of
binding stability
of the complete M
protein
and the truncated Mproteins
complexed with vesicles. In these
experiments
pTM-OM79,
pTM(M1-74),andpTM(M51-229)were
expressed
in theTnT coupledtranscription-translationsystem,andproteins M1-229,
M1-74,
andM51-229,
respectively,
wereheavily
labeled with[35S]methionine. After
purification,
these 35S-labeled Mpro-teinswereincubatedwithDPPC/DPPGvesiclesinthe salt-free
Tricine buffer described in Materials and Methods. These
100-10
e-
<.0
0z
0M-
80-=O.
60--I 40
0. 20-
I
on November 9, 2019 by guest
http://jvi.asm.org/
vesicles, complexed with whole M protein or with the two
truncated M proteins, weredialyzed overnight againstTricine
bufferwithout saltorwith 0.65MNaCl. Thevesicleswerethen
purified byisopycnic centrifugation banding, and the levels of
5S-Mprotein associated with vesicles exposed to no NaClor
0.65 M NaCl were compared by radioactivity determined by
scintillation spectrometry. The results revealed that, after
exposure to 0.65 M NaCl, 83% of the whole M protein and 78% of the M protein truncated to amino acids 51 to 229
remained associatedwith vesicles, respectively.Bycomparison,
only 12% of the M protein truncated to amino acids 1 to 74
remained associated with vesicles exposed to 0.65 M NaCl
(data not shown). These results are quite consistent with the
effectofsaltonthe binding ofMprotein and its truncationsto
vesicles(Fig. 6). These data furthersupportthe hypothesisthat
the N-terminal region of M protein binds to membrane
electrostatically and that theC-terminal region, possibly amino
acids 75to 106,binds tomembrane more stablyby
hydropho-bic bonds.
DISCUSSION
It isgenerally assumed that the VSV M protein is essential
forvirion assemblyinservingasthebridge thatjoins the RNP
to the G-protein-containing plasma membrane. M protein
appearstobeassociated withRNPcores, asjudged by electron
microscopic immunogold labeling (2, 20), aswell asby
chem-icalcross-linkingstudies (9, 17). TheMprotein alsoappearsto
linetheinnersurface ofthevirion membrane in close
proxim-ity to the integral membrane G protein (9), the C-terminal
region ofwhich hasbeen considered to be adocking site for
the M protein-RNP complex (15). However,
plasmid-ex-pressed M protein, in the absence of other viral proteins, is
transportedtoand bindstoplasma membrane (3, 7), and it is
capable on its own of causing evagination of plasma
mem-brane, resultinginbudded vesicles (13). Intact M protein and truncatedMproteins interactwith and bindtoisolated plasma membranes (8) and to phospholipid vesicles (21, 27). In a
reconstitutionsystemtheMproteinshows considerable affinity
forbindingRNPcores(28). The exactRNP-bindingsite(s) on
VSV Mprotein is not known,but studieswith tsmutantsand revertants, aswell as studies using protease protection of M
protein complexed with RNP cores, indicated that
three-dimensional structures markedly influence the stability of
endogenous M protein bound to RNP cores isolated from
virions (10). Recent studies revealed that plasma membrane-associated intactM proteinbinds to RNPcores ina
reconsti-tution assay, but truncated M protein missing the first 14
N-terminal amino acids does notbind RNP cores, suggesting
that the RNP-binding site is located at the N terminus of M
protein (8).Inunpublished studiesusing M proteinsexpressed
by the constructs described in this paper, it was found that
RNPcoresfreeof endogenousMproteinwereabletobindM
proteins truncated to the first 74 amino acids (M1-74) and
missing thefirst 50 aminoacids(M51-229) but thatthey could
bind these somewhat less efficiently than they could bind
complete M protein (M1-229), indicating the possible
exis-tenceoftwositesonMprotein capable ofbinding RNPcores (30).
Evidencethat theMprotein bindstomembranes and liesin
closeproximitytothe VSV virion envelopewasobtainedwith
protein-phospholipidcross-linkingreagents(32).Similar
cross-linkingstudies revealed that a membrane-binding site onthe
virion Mproteinwaslocalizedtoits first19N-terminal amino acids(12). Invitro reconstitution studiesrevealed that soluble
Mproteinbindstosonicated unilamellarphospholipidvesicles
but that it does so far more efficiently if the vesicles contain
negatively charged phospholipids; binding of M protein to
vesicles with acidic phospholipids was markedly reduced by
derivatization of lysine residues or by the presence of 0.5 M
NaCl, indicating the occurrence of primarily electrostatic
binding to the lysine-rich N terminus of the M protein (31).
However, subsequent studies showed thatremoval of the first
43 N-terminal amino acids by trypsin resultedin a truncated M
protein that retained vesicle-binding activity but at
consider-ably reduced efficiency (21), suggesting the presence of two
independent membrane-binding sites on the VSV Mprotein.
About 90% of the plasmid-expressed M protein in transfected
cells was distributed in cytosol, and 10% wastightly bound to
plasma membrane, not dissociable except by detergents (7).
Expression in transfected cells of a truncated M protein
lacking the first 19 amino acids also migrated to plasma
membrane, but this protein was less tightly bound than was
intact M protein; removal of the carboxy-terminal 14 amino
acids has no effect on membrane or nucleocapsid binding of
this truncated M protein (8).
The studies reported here confirm the theory that intracel-lularly synthesized wt M protein migrates to and binds to plasma membrane of cells infected with wt VSV or transfected with a recombinant plasmid expressing only wt M protein. Transfecting deletion mutants revealed that expressed M
proteins truncated to the first 74 amino acids or to aminoacids
51 to229 bind to plasma membrane as determined by indirect
fluorescent-antibody staining or by cell fractionation. Most importantly, deletion of amino acids 1 to 50 and 75 to 106
results in an expressed M protein that remains in the cytosol
and cannot be detected in plasma membrane either by
fluo-rescent-antibody staining or by cell fractionation. These results
collectively indicate the presence of two membrane-binding
sites on wt Mprotein, one at the N terminus in the
neighbor-hoodof amino acids 1 to 50 and a second between amino acids
75 and 106. These in vivo studies were confirmed by in vitro
experiments in which the entire M protein and its three deletion mutants were tested for their capacity to bind
soni-cated phospholipid vesicles. The double-deletion mutant
(M51-74/107-229)showed only background binding to vesicles
even in the absence of salt or in a 0.15 M NaCl buffer. The
vesicle binding exhibited by the two single-deletion mutants (M1-74 and M51-229) was somewhat less than that exhibited
by the complete M protein (M1-229) at physiological salt
concentrations. The M protein truncated to the first 74 amino
acids (M1-74) did not bind to vesicles beyond the background
level in areaction buffer containing 0.65 M NaCl, whereas the
M protein deleted of the first 50 amino acids (M51-229)
showed nosignificant decline in its capacity to bind vesicles at
high saltconcentrations. Moreover, after prebinding to vesicles
in alow-saltenvironment, truncated protein M1-74 was readily
dissociated from vesicles by exposure to 0.65 M NaCl, whereas
truncated protein M51-229 remained stably associated with
vesiclesexposed to 0.65 M NaCl. These data appear to confirm
the presence of two membrane-binding sites on VSV M
protein. (i) The N-terminal site, rich in charged amino acids,
appears to bind to cellular or vesicle membranes by
electro-static forces. (ii) The region on M protein spanning amino
acids 75 to 106 isrich inhydrophobic amino acids and is stably
bound to cellular or vesicle membranes in an integrative manner. Quite clearly, the loss of membrane-binding activity of
thedouble-deletion mutant could be due to a conformational
change and to aberrant folding of the M protein, folding which
was more extensive than that which occurred in the case of
each ofthe single deletions alone.
The M protein synthesized at the nonpermissive
on November 9, 2019 by guest
http://jvi.asm.org/
tureincells infected with the VSVmutanttsO23wasalsonot
detectable, or barelydetectable, in plasma membrane either by
fluorescent-antibody staining or by cell fractionation. This
mutant has amino acid substitutions at residues 21, 111, and
227 (18). On the basis of studies with spontaneous revertants,
the major mutation resulting in markedly reduced assembly
and yield of infectious progeny atthe nonpermissive
temper-ature is a leucine--phenylalanine substitution at amino acid
111 (18); supporting this conclusion was the finding that
expression ofmutant Mprotein withaPhe-*Leureversionat
residue 111 complements growth ofts023 at the
nonpermis-sive temperature (14). Studies with similar M-gene mutants
and revertants led Kaptur et al. (10) to conclude that the temperature-sensitive phenotype affected the RNP-binding
affinity ofMprotein because of altered conformation. Itseems
entirely likelythat thereducedbindingtoplasmamembrane of
the tsO23 mutant M protein in experiments reported here is
alsodue to altered conformation.
Another property of the VSV M protein is its capacity to
cause acytopathic effect evidencedbycellrounding, probably
due to dissolution of actin filaments, in cells infected withwt
VSV or its mutant tsG33 at permissive temperatures (5).
Another, probably unrelated, effect of M protein is the
inhibi-tion of cell-directed transcription of target genes (4)whenthe
M protein is transported to the nucleus (16). Cells infected
with the VSV M-genemutanttsG33 atthe restrictive
temper-aturedonot exhibit thecytopathic effect of cellrounding (5).
We haveobtained, asreported here, identical results with the
VSV M-gene mutant ts023, which causes cell rounding and
actinfilament dissolutionatpermissivetemperaturesbutnot at
nonpermissive temperatures. It seems entirely likely that the
cell-rounding effect also depends on correct conformation of
the Mprotein onlyatpermissivetemperatures.Wefound that
expression ofwt M protein by pTM-OM79 also results in cell
rounding and actin filament dissolution,whereas none of the
threedeletionmutantMproteinscaused cell
rounding
oractinfilament dissolution. It seems likelythat alteredconformation
of ts023 mutant M protein produced at the
nonpermissive
temperature or caused by deletion mutants compromises its
capacitytoproduce thiscytopathiceffect in infected or
trans-fected cells. Itis temptingto
speculate
that the sameconfor-mation of wt, butnotmutant,Mproteinisresponsiblefor both
theaffinityfor membranebinding and the
cell-rounding
cyto-pathic effect. Conceivably, intracellularly
synthesized
Mpro-tein in thecorrect conformationbinds tothe inner surface of
the plasma membrane, resulting in detachment of actin
fila-mentsand subsequentloss of normalpolygonal cell
morphol-ogy.
The datapresented here, and thosepresented
previously by
us and others, suggest a possible model testable
by
futureexperimentation. Incells infected withwtVSV, mostoftheM protein attaches to and tightly coils newly
synthesized
RNPcores, which eventually migrate and bind to virus-converted
plasmamembrane. The remainder of theM
protein
migrates
independently and binds to plasma membrane by a route
different fromthat of Gprotein
(7).
TheMprotein, aloneorcomplexed
with RNP cores, bindstothe inner surface of theplasma membrane by electrostatic forces
generated by
thepositively charged N terminus of the M
protein
and thenegativelycharged headgroupofphosphatidylserine
(27).
Thiselectrostatic binding possibly results in a conformational
changethat exposes the
hydrophobic
domain ofMprotein
in theregionof amino acids 88to119,which then couldpartition
into thelipid bilayer, formingastable
complex
notdissociableby high-ionic-strength buffers. The stable M
protein-plasma
membrane complex conceivably detaches the ends of actin
filaments, resulting
in the loss of the characteristicpolygonal
shape
and in thesubsequent rounding
of the cell. The finalsteps of
envelopment
of RNP-Mprotein
coresby
theG-protein-containing plasma membrane and
budding
of theprogeny virionsare
beyond
the scope of this paper.ACKNOWLEDGMENT
This research was supported by Public Health Service grant R37 AI-11112from the National Institute ofAllergyand Infectious Dis-eases.
REFERENCES
1. Barenholz,Y., N.J. Moore,andR. R.Wagner. 1976.Enveloped
viruses as model membrane systems: microviscosity ofvesicular stomatitis virus and host cell membranes.Biochemistry 15:3563-3570.
2. Barge, A., Y. Gaudin, P. Coulon, and W. H. Ruigrok. 1993. Vesicular stomatitis virus Mproteinmaybeinsidethe
ribonucleo-capsidcoil.J. Virol.67:7246-7253.
3. Bergmann, J.E.,and P.J.Fusco.1988.The Mproteinof vesicular stomatitis virus associatesspecificallywith the basolateral
mem-branes ofpolarizedepithelialcellsindependentlyofthe Gprotein.
J.Cell Biol. 107:1707-1715.
4. Black, B. L., and D. S. Lyles. 1992. Vesicular stomatitis virus matrix protein inhibits host cell-directed transcription oftarget genes in vivo. J. Virol. 66:4058-4064.
5. Blondel, D., G. G. Harmisom, and M. Schubert. 1990. Role of
matrixproteinincytopathogenesisof vesicularstomatitisvirus. J. Virol.64:1716-1725.
6. Carroll,A.R.,and R. R.Wagner.1979. Role of themembrane(M)
protein in endogenous inhibition of in vitro transcription of
vesicularstomatitisvirus. J. Virol. 29:134-142.
7. Chong, L. D., and J. K. Rose. 1993. Membrane association of
functional vesicular stomatitis virus matrixproteinin vivo.J. Virol. 67:407-414.
8. Chong, L.D., andJ. K. Rose. 1994. Interaction of normal and mutantvesicular stomatitis virus matrixproteinswiththeplasma
membrane andnucleocapsids.J.Virol. 68:441-447.
9. Dubovi,E.J.,and R.R.Wagner.1977.Spatialrelationshipsof the
proteins of vesicular stomatitis virus: induction of reversible
oligomers by cleavable protein cross-linkers and oxidation. J.
Virol.22:500-509.
10. Kaptur,P.E.,R. B. Rhodes,andD.S.Lyles.1991.Sequencesof thevesicularstomatitis virus matrixproteininvolved inbindingto
nucleocapsids.J.Virol. 65:1057-1065.
11. Kyte,J.,and R. F. Doolittle.1982.Asimplemethod fordisplaying
thehydropathiccharacter ofaprotein.J. Mol.Biol. 157:105-132.
12. Lenard, J., and R. Vanderoef. 1990. Localization of the
mem-brane-associatedregionofvesicularstomatitisvirusM
protein
at the N terminus, using the hydrophobic, photoreactive probe'251-TID.
J. Virol. 64:3486-3491.13. Li,Y.,L.Luo,M.Schubert,R. R.Wagner,and C.-Y.Kang. 1993.
Viralliposomesreleased from insect cells infected with recombi-nantbaculovirusexpressingthe Mproteinofvesicular stomatitis virus. J.Virol. 67:4415-4420.
14. Li,Y., L. Luo, and R.R. Wagner. 1989.
Transcription
inhibition site on the M protein of vesicular stomatitis virus locatedby
marker rescue of mutant tsO23(III) with M-gene
expression
vectors.J. Virol. 63:2841-2843.
15. Lyles,D. S.,M.McKenzie, andW. Parce. 1992. Subunit
interac-tions ofvesicular stomatitisvirusenvelope
glycoprotein
stabilizedbybindingtoviralmatrix
protein.
J.Virol. 66:349-358.16. Lyles, D. S., L. Puddington, and B. J. McCreedy, Jr. 1988.
Vesicular stomatitis virus Mproteininthe nuclei of infected cells. J.Virol. 62:4387-4392.
17. Mancarella,D.A., andJ. Lenard. 1981.Interaction of
wild-type
and mutant M protein of vesicular stomatitis virus with viral
nucleocapsid andenvelope inintact virions. Evidence from [125]1 iodonaphthylazidelabelingand
specific
cross-linking.
Biochemis-try 20:6872-6877.18. Morita,J.,R.Vanderoef,andJ.Lenard. 1987.
Phenotypic
rever-tants of temperature-sensitive M
protein
mutants of vesicularon November 9, 2019 by guest
http://jvi.asm.org/
stomatitis virus: sequenceanalysis andfunctional characterization. J.Virol.61:256-263.
19. Moss, B., 0. Elroy-Stein, T. Mizukami, W. A. Alexander, and T. R. Fuerst. 1990. New mammalian vector. Nature (London) 348:91-92.
20. Newcomb, W. W., and J. C. Brown. 1981. Role of the vesicular stomatitisvirus matrixprotein in maintainingthe viral nucleocap-sid in the condensed form found in nativevirions. J. Virol. 39: 295-299.
21. Ogden, J. O., R.Pal, and R. R. Wagner. 1986.Mapping regions of the matrix protein of vesicular stomatitis virus which bind to ribonucleocapsids,liposomes, and monoclonal antibodies. J. Virol. 58:860-868.
21a.Ono,K., M. E.Dubois-Dalcq, M. Schubert, and R. A. Lazzarini. 1987. Amutated membraneprotein of vesicular stomatitis virus hasanabnormal distribution within theinfected celland causes defective budding.J.Virol. 61:1332-1341.
22. Pal, R., and R. R. Wagner. 1987. Rhabdovirus membrane and maturation, p. 75-128. In R. R. Wagner(ed.),Therhabdoviruses. PlenumPublishingCorp., New York.
23. Rose, J. K., and C. J. Gallione. 1981.Nucleotidesequencesofthe mRNAsencoding the vesicularstomatitisvirus Gand Mproteins determined from nucleotide sequencescontaining the complete coding regions.J.Virol.39:519-528.
24. Shipley, J. B., R. Pal, and R. R. Wagner. 1988. Antigenicity, function,and conformation ofsyntheticoligopeptide correspond-ing to amino-terminal sequencesof wild-type and mutant matrix proteins of vesicular stomatitis virus. J. Virol. 62:2569-2577.
25. Thomas, D., W. W. Newcomb, J. C. Brown, J. S. Wall, J. E. Hainfeld, B. L. Trus, and A. D. Stevens. 1985. Mass and molecular composition of vesicularstomatitis virus: ascanningtransmission electronmicroscopy analysis.J. Virol. 54:598-607.
26. Wagner, R. R. 1987. Rhabdovirus biology and infection: an overview, p. 9-74. In R. R. Wagner (ed.), The rhabdoviruses. Plenum Publishing Corp., New York.
27. Wiener, J. R., R. Pal, Y. Barenholz, and R. R. Wagner. 1985. Effect of the vesicular stomatitis virus matrix protein on the lateral organizationoflipid bilayers containing phosphatidylglycerol:use of fluorescent phospholipid analogues. Biochemistry 24:7651-7658.
28. Wilson, T., and J. Lenard. 1981. Interaction of wild-type and mutant Mproteinofvesicularstomatitis virus withnucleocapsids in vitro.Biochemistry20:1349-1354.
29. Ye, Z., R. Pal, J. W. Fox, and R. R. Wagner. 1987. Functional and antigenicdomainsof the matrix(Ml)protein of influenzaAvirus. J. Virol. 61:239-246.
30. Ye, Z., W.Sun, and R. R. Wagner. Unpublished data.
31. Zakowski, J. J., W. A. Petri, Jr., and R. R. Wagner. 1981. Role of matrix protein inassemblingthe membrane of vesicularstomatitis virus: reconstitution of matrix protein with negatively charged phospholipid vesicles.Biochemistry 20:3902-3907.
32. Zakowski, J. J., and R. R. Wagner. 1980. Localization of mem-brane-associated proteins in vesicular stomatitis virus byuse of hydrophobic membrane probes and cross-linking reagents. J. Virol. 36:93-102.